
Cumplimiento normativo en el marco del registro y la regulación de « Dispositivos Médicos » en la UE
La Unión Europea representa uno de los mercados Dispositivos Médicos más regulados y comercialmente atractivos del mundo. Junto con los 27 member states, los 3 EEA países y Turquía, el mercado de la Unión funciona bajo un ecosistema normativo armonizado definido por el Reglamento (UE) 2017/745 sobre productos sanitarios ( EU MDR ) y el Reglamento (UE) 2017/746 sobre diagnósticos in vitro (EU IVDR) (2017/746); de este modo, la región garantiza normas uniformes de seguridad y rendimiento en todas las categorías de productos. Para comercializar legalmente un producto en el mercado de la Unión Europea, los fabricantes deben obtener el marcado CE, que confirma el cumplimiento de la normativa aplicable en función de la clasificación de riesgo del producto y la vía de evaluación de la conformidad. El incumplimiento de estos requisitos puede acarrear graves consecuencias, como la retirada del producto, la incautación en aduana, la suspensión de la certificación o la pérdida total del acceso al mercado.
Según el régimen normativo vigente:
- El marcado CE es obligatorio para todos los productos sanitarios y los productos para diagnóstico in vitro antes de su comercialización.
- El registro de productos sanitarios en EUDAMED se está implantando por fases, y ya hay varios módulos activos cuyo uso es obligatorio.
- El nombramiento de un representante autorizado europeo (EAR) es obligatorio para cualquier fabricante con sede fuera de la UE,EEA o Turquía.
- Las normas de clasificación más estrictas del MDR y el IVDR exigen una documentación técnica sólida, pruebas clínicas o de rendimiento, y un seguimiento continuo a lo largo del ciclo de vida.
Freyr apoya a los fabricantes a lo largo de todo este proceso, facilitándoles una entrada fluida y conforme a la normativa en el mercado de la Unión. Desde el desarrollo de una estrategia de marcado CE y la elaboración de la documentación técnica hasta la gestión de las solicitudes ante los organismos notificados y la actuación como su representante autorizado en la UE, Freyr garantiza el pleno cumplimiento de los reglamentos MDR e IVDR, así como un acceso sin obstáculos a todas las regiones de la UE member states, EEA y a Turquía.
Proceso paso a paso para el cumplimiento de la normativa de la UE
El acceso al mercado de un dispositivo de diagnóstico in vitro ( Dispositivos Médicos , IVD) en la UE implica varias fases bien definidas. A continuación te explicamos cómo Freyr gestiona todo el proceso por ti:
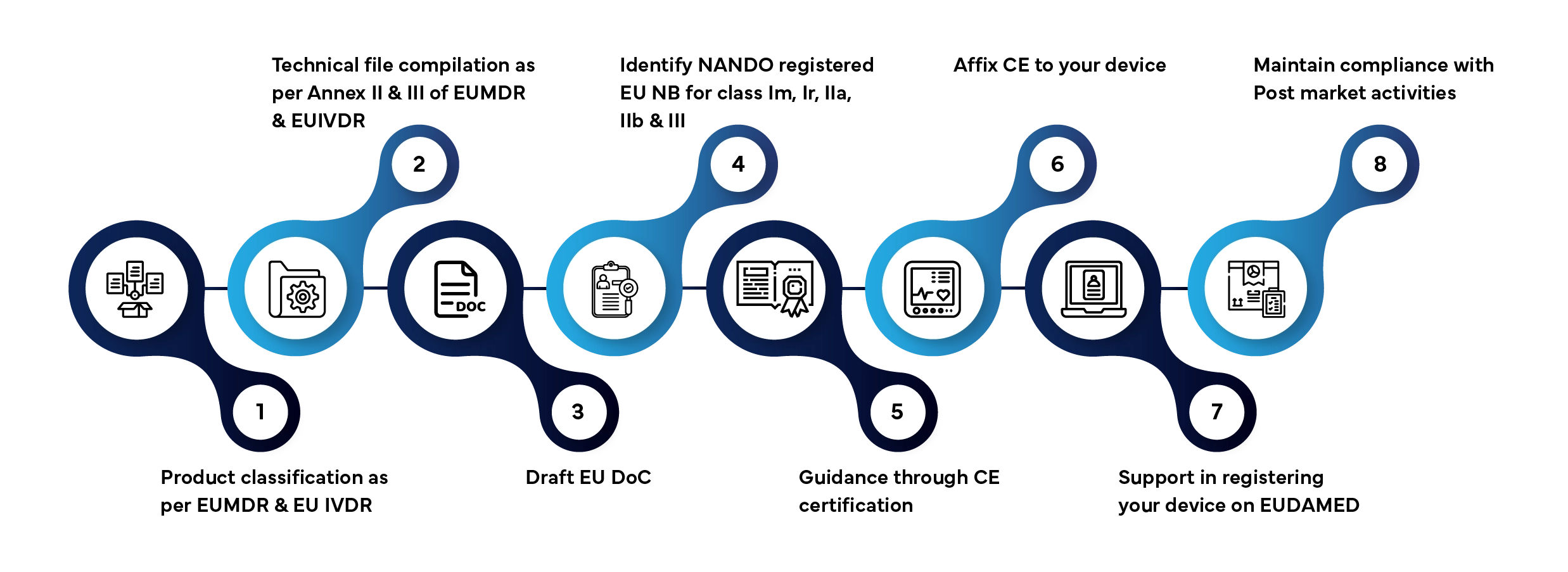
Plazo de tramitación habitual: de 3 a 12 meses, en función de la clase de dispositivo y de si la documentación está completa.
Freyr Dispositivos Médicos Ofertas clave en la UE
- Estrategia regulatoria y transición al MDR/IVDR: elaboramos hojas de ruta regulatorias end-to-end adaptadas al Reglamento sobre dispositivos médicos ( EU MDR ) y al Reglamento de la UE sobre productos para diagnóstico in vitro (IVDR), garantizando una transición fluida desde los regímenes de las directivas y la adaptación a los requisitos actuales de la UE.
- Documentación técnica y evaluación de la conformidad: nuestro equipo le ayuda en la elaboración, revisión y presentación de sus expedientes técnicos y de diseño, le asiste en las gestiones con los organismos notificados y se encarga de gestionar el marcado CE, así como de ofrecer apoyo en las pruebas de seguridad y rendimiento de los productos sanitarios y en los procesos de conformidad.
- Evaluación clínica y de rendimiento: Freyr ofrece servicios especializados de elaboración de informes de evaluación comparativa (CER), informes de evaluación de rendimiento (PER), planes de seguimiento de la seguridad ( PMCF) y planes de seguimiento de la seguridad y la eficacia (PMPF), informes periódicos de seguridad (PSUR), informes de variación significativa (SVR), informes de seguimiento de la seguridad (CPR) para productos de diagnóstico in vitro (IVD), documentación de riesgos y contenidos de evaluación biológica, garantizando la claridad técnica y la precisión normativa en todas las clases de productos sanitarios.
- Asistencia para el registro en UDI y EUDAMED: nos aseguramos de que su sistema de identificación única de productos sanitarios (UDI) cumpla con la normativa y le ayudamos con el registro en la base de datos europea de productos sanitarios EUDAMED, así como con la gestión asociada de su ciclo de vida.
- Representante autorizado europeo (EAR) y representación local: para los fabricantes fuera de la UE,EEA y Turquía, actuamos como su representante autorizado europeo (EAR) designado y ofrecemos asistencia local en materia de cumplimiento normativo en toda la UE member states.
- Vigilancia poscomercialización (PMS): Freyr le ayuda a establecer, implementar y mantener sistemas de PMS, incluidos el Plan de Vigilancia Poscomercialización (PMSP), el informe de vigilancia poscomercialización (PMSR), el informe periódico de actualización de seguridad (PSUR), los planes de seguimiento clínico y de rendimiento poscomercialización (PMCF/PMPF), la notificación de casos de vigilancia, las FSCAs, las FSNs y las CAPAs, garantizando así el mantenimiento continuo del marcado CE y un acceso sostenible al mercado, con el cumplimiento normativo durante todo el ciclo de vida del producto.
- Registro de productos con marcado CE: Freyr gestiona todo el proceso de registro para la obtención del marcado CE de la UE, prestando apoyo en la evaluación de la conformidad, elaborando la documentación necesaria para la presentación de solicitudes, colaborando con los organismos notificados y garantizando la obtención de las autorizaciones en los plazos previstos para todas las categorías de productos de diagnóstico in vitro ( Dispositivos Médicos ) y de dispositivos médicos (IVD).
- Asistencia en sistemas de gestión de la calidad: Ofrecemos asistencia para la implantación y el mantenimiento de sistemas de gestión de la calidad que cumplan con la norma ISO 13485, en consonancia con los requisitos de calidad y seguridad de la Directiva sobre productos sanitarios ( EU MDR) y el Reglamento sobre dispositivos médicos (IVDR), así como con las expectativas de los organismos notificados.
- Cumplimiento normativo en materia de etiquetado: Nuestro equipo se asegura de que el etiquetado, las instrucciones de uso, el embalaje y los símbolos cumplan con los requisitos del MDR/IVDR y con la normativa multilingüe de la UE, garantizando la coherencia y el cumplimiento normativo en los 27 Estados miembros de la UE member states.
Oferta de servicios del representante autorizado en la UE (EAR)

Registro de dispositivos ante las autoridades de la UE
Para los fabricantes no pertenecientes a la UE, Freyr actúa como representante autorizado en la UE (EAR) designado, de conformidad con los requisitos del MDR/IVDR. Como entidad con sede en Alemania, Freyr presta apoyo en las actividades de registro de productos ante la autoridad competente alemana correspondiente y mantiene los registros reglamentarios exigidos. Para otros e Member States es de la UE, Freyr ofrece apoyo en materia de normativa, según proceda, con el fin de garantizar el cumplimiento de los requisitos aplicables a los productos y facilitar su comercialización en el mercado de la Unión.

Documentación y garantía de conformidad
Nuestros expertos en normativa comprueban que su Declaración de conformidad (DoC), los certificados CE y los expedientes técnicos estén completos, actualizados y cumplan con el MDR y el IVDR. Freyr garantiza que todo esté perfectamente preparado para la evaluación de la conformidad y la presentación de las solicitudes de marcado CE.

Respuesta a las consultas de las autoridades competentes
Si fuera necesario, Freyr se encarga, en su nombre, de toda la comunicación directa y de las solicitudes de aclaración por parte de las autoridades competentes de la UE o de los organismos notificados, garantizando respuestas puntuales y precisas y contribuyendo a evitar retrasos en las autorizaciones o en las revisiones reglamentarias posteriores a la comercialización.

Vigilancia y comunicación de incidentes
En su calidad de EAR, Freyr actúa como principal enlace para las comunicaciones relacionadas con la seguridad. Cuando sea necesario, coordinamos las notificaciones de incidentes, las medidas correctivas de seguridad sobre el terreno (FSCA) y los informes de vigilancia entre el fabricante, los profesionales sanitarios y las autoridades, con el fin de garantizar una respuesta adecuada y el cumplimiento de la normativa.

Preparación para inspecciones y auditorías
Freyr conserva toda la documentación, la correspondencia y los registros obligatorios para las auditorías e inspecciones de las autoridades. Nuestro equipo se asegura de que la documentación técnica, el etiquetado y los registros posteriores a la comercialización estén fácilmente disponibles y cumplan plenamente con los requisitos del MDR y el IVDR.
¿Por qué asociarse con Freyr?
- End-to-end experiencia en materia de normativa que abarca desde el registro previo a la comercialización hasta la vigilancia posterior a la comercialización, gestionando todas las fases del cumplimiento normativo.
- Una trayectoria contrastada con más de 2.000 registros de dispositivos completados con éxito en diversas categorías.
- Una sólida presencia en la UE con sede en Alemania (EAR), complementada por especialistas en normativa sobre el terreno en Reading y respaldada por los equipos de ejecución globales de Freyr.
- Una planificación de la transición a medida que ofrece apoyo estratégico para obtener la certificación CE de forma fluida y rentable.
- Equipos de entrega multirregionales con apoyo sobre el terreno a través de las operaciones de Freyr en la UE, con sede en Alemania.
- Experiencia demostrada en la adaptación a los reglamentos MDR e IVDR y en la transición de dispositivos heredados
- Una gestión transparente del proyecto con comunicación directa con el organismo notificado

Celebrando el éxito de los clientes
Dispositivos Médicos
Registro y soporte de LR
Global
Freyr ha sido un socio indispensable para lograr una rápida escalabilidad global para nuestro negocio de Software como Dispositivo Médico (SaMD). Como startup, adquirir experiencia en regulaciones mundiales es prohibitivamente caro. Los precios competitivos y los servicios personalizados de Freyr nos permitieron obtener esa experiencia a una fracción del costo de los recursos a tiempo completo. La capacidad de respuesta y adaptabilidad de su equipo a las prioridades del proyecto han facilitado enormemente nuestro progreso. Recomendamos Freyr a cualquier empresa que busque orientación y apoyo experto en el ámbito reglamentario de los Dispositivos Médicos.
Arie Henkin
Vicepresidente - Calidad y Asuntos Reglamentarios, con sede en Australia, Empresa líder en SaMD
Dispositivos Médicos
Servicios de Representación Suizos
Japón y Suiza
Realmente disfruto mi tiempo trabajando con Freyr, y los considero un activo verdaderamente valioso y una extensión de mi propio equipo. Son confiables y precisos, y sus precios son competitivos. Además, no dudaré en colaborar con Freyr de nuevo.
Darren Mansell
Gerente de Asuntos Regulatorios, con sede en el Reino Unido, Empresa Global de Diseño y Fabricación de Dispositivos Médicos
Dispositivos Médicos
Servicios de Registro y AR
Malasia e Indonesia
Freyr ofrece un servicio fiable con experiencia en muchos países. Puedo confiar en Freyr para obtener la información necesaria y tomar una decisión informada antes de firmar un acuerdo formal de alcance de trabajo. Una vez que un proyecto está en marcha, el equipo de Freyr actúa profesionalmente para ejecutar el trabajo con una excelente comunicación del progreso.







