Registro de Diagnóstico In Vitro (IVD) - Descripción general
En el panorama en constante cambio de la tecnología sanitaria, los Dispositivos Médicos desempeñan un papel fundamental en la mejora de la atención al paciente, los diagnósticos y los resultados de los tratamientos. Se espera que el mercado global de Dispositivos Médicos crezca de 536.120 millones de USD en 2023 a 799.670 millones de USD para 2030, con una Tasa de Crecimiento Anual Compuesto (CAGR) proyectada del 5,5 %. Este crecimiento se impulsa por el creciente número de ingresos hospitalarios, el aumento de los procedimientos quirúrgicos y diagnósticos, y la creciente demanda de Dispositivos Médicos tanto en mercados desarrollados como emergentes. Los principales actores de la industria también están aumentando sus inversiones en investigación y desarrollo para crear equipos avanzados que satisfagan la demanda de innovación, impulsando aún más la cuota de mercado.
Escenario reglamentario global para el registro de Dispositivos Médicos
Las reglamentaciones de Dispositivos Médicos varían según el país, lo que hace esencial que los fabricantes comprendan y manejen estas diferencias para ingresar a los mercados globales. Los esfuerzos para armonizar las reglamentaciones y promover la colaboración internacional tienen como objetivo simplificar este proceso y mejorar la seguridad del paciente a escala global. Cada país tiene su propio conjunto único de reglamentaciones y requisitos, que están influenciados por factores como los niveles de riesgo, las declaraciones de productos y el uso previsto. Algunas de las directrices destacadas disponibles para el registro de Dispositivos Médicos a nivel mundial incluyen la FDA (US), las normas ISO, el Grupo de Trabajo de Armonización Global (GHTF), la Organización Mundial de la Salud (WHO) y la Unión Europea (UE). También deben estar preparados para renovar su registro anualmente y cumplir con las expectativas de las Autoridades Reglamentarias.
El registro de Dispositivos Médicos en los mercados internacionales requiere un enfoque personalizado, que implica una estrecha colaboración con las Agencias de Salud pertinentes para su aprobación. El proceso típico para el registro de Dispositivos Médicos implica los siguientes pasos:
- Evaluar si un dispositivo específico cumple los criterios para su clasificación como Dispositivo Médico.
- Clasificación de los dispositivos según los riesgos asociados.
- Identificación de las normas pertinentes y los requisitos de datos especificados por la Agencia de Salud correspondiente.
- Generación de los datos necesarios según lo exigido por la Agencia.
- Compilación de un expediente técnico de acuerdo con los requisitos específicos de cada país.
- Presentación de la solicitud y resolución de cualquier consulta o preocupación hasta la obtención de la aprobación.
- Gestión del ciclo de vida del dispositivo después de la aprobación.
Nuestras Competencias
- Análisis de riesgo inicial
- Investigación de mercado - Conocimientos de mercado específicos del producto
- Ampliación de personal
- Estrategia reglamentaria preliminar.
- Mercados y vías potenciales.
- Expediente de diseño y análisis de riesgos.
- Sistema de Gestión de Calidad (SGC) ISO 13485
- Programa de Auditoría Única de Dispositivos Médicos (MDSAP)
- Preevaluación de QMS ISO 13485
- Estrategia reglamentaria
- Freyr IMPACT (Plataforma de Inteligencia Reglamentaria)
- Verificación y validación del diseño
- Gestión de riesgos
- Elaboración de documentación técnica
- Estrategia reglamentaria
- Requisitos reglamentarios
- Herramienta rDMS de Freyr (Sistema de Gestión de Datos/Documentación)
- Validación de procesos y clínica
- Etiquetado y Artwork final
- Representación en el país
- Presentación reglamentaria
- El marcado de Conformidad Europea (CE) de la Unión Europea (UE) y el marcado de Evaluación de Conformidad del Reino Unido (UKCA)
- Certificación de acceso al mercado global
- Apoyo para auditorías de Organismos Notificados (ON)/Organismos Aprobados
- Representación en el país
- Aprobaciones reglamentarias
- Vigilancia poscomercialización (PMS)
- Seguimiento Clínico Post-comercialización (PMCF)
- Mantenimiento anual del expediente técnico (Informe de Evaluación Clínica (CER)/Gestión de Riesgos)
- Renovaciones reglamentarias
- Lanzamientos de nuevos productos al mercado
- Comunicación con la Autoridad Competente/Organismo Notificado/Aprobado
- Soluciones automatizadas de Farmacovigilancia (PV)
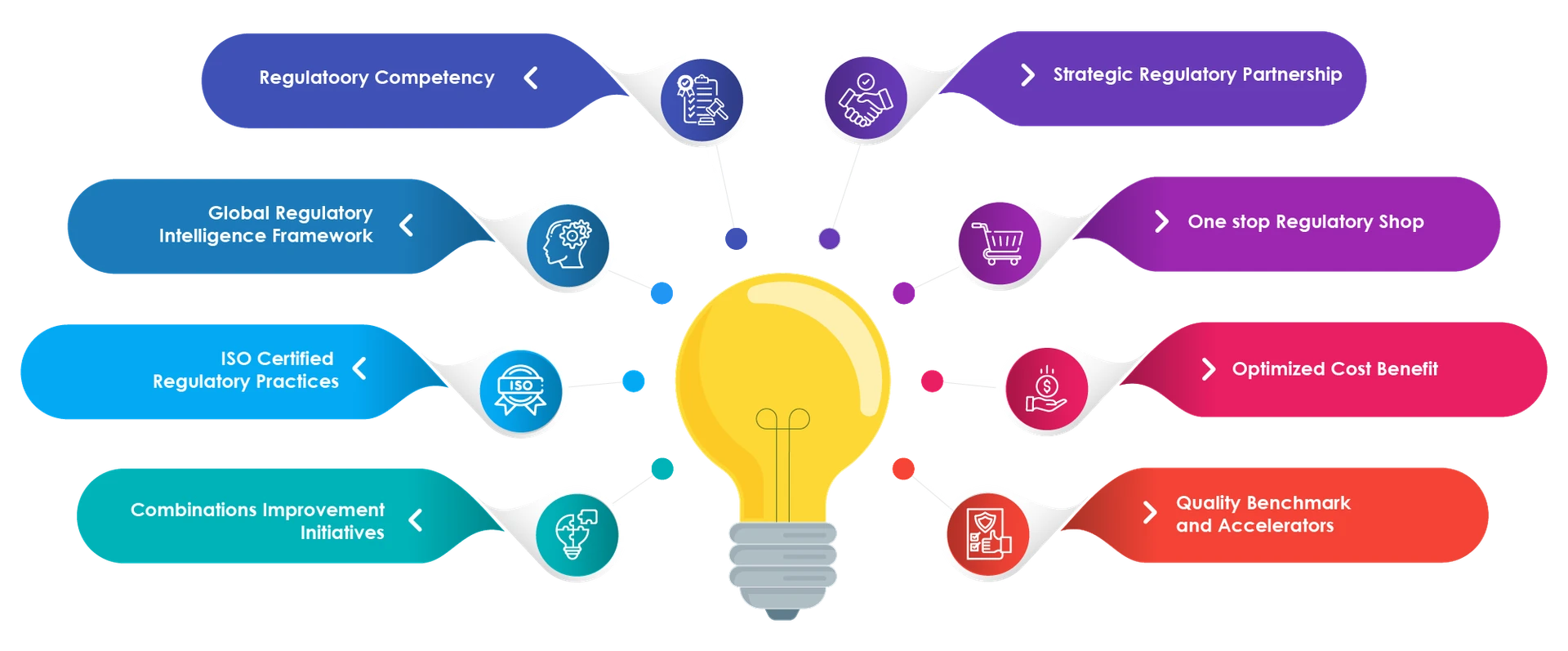
¿Por qué Freyr?
Registro de Dispositivos Médicos
- Estrategia reglamentaria integral para IVD.
- Soporte reglamentario para documentos de desarrollo de productos, como los Expedientes de Historial de Diseño (DHFs).
- Estrategia de cumplimiento de QMS.
- Cumplimiento reglamentario, análisis de brechas y subsanación de documentos técnicos y sistemas de calidad.
- Servicios de etiquetado reglamentario y redacción técnica.
- Servicios de inteligencia reglamentaria y de mercado.
- Servicios de traducción de documentos y etiquetado.
- Servicios de enlace con agencias de salud.
- Servicios de Artwork reglamentario.
- Servicios de Farmacovigilancia y PMS.
- Servicios de publicación.
- Servicios de redacción médica.

- Presentaciones exitosas para diversas clases de IVD.
- Personal dedicado y experto para proporcionar soporte reglamentario para Dispositivos Médicos e IVD.
- Entrega puntual de los resultados.
- Acceso a filiales locales para afrontar los desafíos de la Autoridad y los requisitos específicos del idioma.
- Soporte de representante local o legal con un modelo rentable.
- Servicios de gestión de recursos reglamentarios/refuerzo de personal.