Resumen del Registro de Dispositivos Médicos de la US FDA
Estados Unidos de América (USA) es conocido por ser un mercado altamente reglamentado para Dispositivos Médicos, con vías y requisitos de registro bien definidos. Las regulaciones iniciales de Dispositivos Médicos de US datan de 1976 y han evolucionado con el tiempo. Están regulados por el Centro de Dispositivos y Salud Radiológica (CDRH) bajo la Administración de Alimentos y Medicamentos (FDA). Freyr Solutions ha ayudado a múltiples fabricantes de dispositivos a cumplir con el proceso de registro de Dispositivos Médicos de la USFDA.
![]()
Autoridad reglamentaria: Administración de Alimentos y Medicamentos (FDA)![]()
Reglamentación: Título 21 del Código de Regulaciones Federales (21 CFR) Partes 800 – 1299![]()
Ruta Reglamentaria: Notificación Pre-Comercialización o Aprobación Pre-Comercialización o Clasificación De Novo![]()
Representante Autorizado: Agente de EE. UU.![]()
Requisito de QMS: Reglamento del Sistema de Calidad (QSR) (21 CFR parte 820)![]()
Evaluación de Datos Técnicos: Centro de Dispositivos y Salud Radiológica![]()
Validez de la licencia: Ilimitada![]()
Requisitos de Etiquetado: 21 CFR Parte 801![]()
Formato de Presentación: Papel y CD/DVD![]()
Idioma: Inglés
Clasificación de Dispositivos Médicos en EE. UU.
La FDA clasifica los Dispositivos Médicos en 3 categorías basadas en el riesgo: Clase I, Clase II y Clase III, donde los dispositivos de Clase I se consideran de bajo riesgo y los de Clase III se asocian con un alto riesgo. Los requisitos de registro y el proceso varían según la clase de dispositivo.
| Clase de Dispositivo | Riesgo | Vía de Registro Para su aprobación |
|---|---|---|
| Yo | Riesgo bajo | Exento de 510(k) |
| II | Riesgo Moderado (Con dispositivo de referencia) | Notificación Pre-Comercialización/510(k) |
Riesgo Moderado (Sin dispositivo de referencia) | Solicitud De Novo | |
| III | Riesgo alto | Aprobación Precomercialización (PMA) |
Agente de la FDA de US
Las empresas sin oficinas locales en los EE. UU. deben designar un Agente de la FDA de EE. UU. para representar al fabricante. El agente de la FDA de EE. UU. debe residir en los EE. UU. o mantener un lugar de negocios en los EE. UU. Las responsabilidades que debe cumplir el agente están predeterminadas por la US FDA como parte de las regulaciones CFR.
Navegue por las Preguntas Frecuentes (FAQ) sobre el Agente de US.
Reuniones interactivas con la FDA de US
La FDA de US apoya a los fabricantes a través de varios tipos de reuniones de Q-Submission para cumplir diferentes objetivos. Estas reuniones con la agencia, antes del inicio o durante el desarrollo del dispositivo, y previas a la presentación de las solicitudes de registro de Dispositivos Médicos de la FDA de US, ayudan a los fabricantes a optimizar los plazos y los costos incurridos para la comercialización del dispositivo.
Registro de Dispositivos Médicos en US
Los dispositivos pueden ser aprobados por el CDRH, FDA a través de cualquiera de las diversas vías de registro. Se listan a continuación:
Dispositivos Médicos de Clase I: Los dispositivos de clase I suelen estar exentos de la presentación de GMP y 510(k) y no requieren aprobación previa de la US FDA para comercializarlos en EE. UU. El fabricante debe cumplir con otros requisitos como el registro del establecimiento, la lista de dispositivos, UDI, PMS, etc.
Dispositivos Médicos de Clase II: Los dispositivos de riesgo medio con dispositivos de referencia aprobados por 510(k) pueden optar por la Notificación Previa a la Comercialización 510(k) (PMN), también llamada registro 510(k). El dispositivo en cuestión deberá establecer Equivalencia Sustancial (SE) con los dispositivos de referencia identificados y declarados. Esta vía es la más ampliamente adaptada para el registro de dispositivos en EE. UU. Los fabricantes de dispositivos de riesgo medio sin dispositivos de referencia pueden solicitar la clasificación por parte de la US FDA a través de solicitudes De-Novo.
Dispositivos Médicos de Clase III: Los fabricantes de dispositivos de Clase III de alto riesgo deben presentar una solicitud de Aprobación Previa a la Comercialización (PMA) a la US FDA. Los dispositivos deben someterse a una evaluación clínica detallada y el fabricante debe presentar datos detallados de seguridad y eficacia de estudios clínicos. La US FDA llevaría a cabo una inspección del SGC como parte de la evaluación antes de emitir una Aprobación Previa a la Comercialización para el dispositivo.
Registros de Dispositivos Médicos No CDRH
Según las indicaciones de uso, algunos productos límite considerados Dispositivos Médicos en otros países, como respiradores quirúrgicos, desinfectantes y productos combinados, involucran a otras agencias, como el Centro para el Control de Enfermedades (CDC), el Instituto Nacional para la Seguridad y Salud Ocupacional (NIOSH), la Environmental Protection Agency (EPA), el Centro de Evaluación e Investigación Biológica (CBER) y el Centro de Evaluación e Investigación de Medicamentos (CDER).
Requisitos de Cumplimiento Post-Aprobación para Dispositivos Médicos
Todos los fabricantes de dispositivos deben cumplir con los siguientes requisitos posteriores a la aprobación:
- Requisito de registro y listado: Los establecimientos de todas las clases de dispositivos deben registrarse en la base de datos FURLs y el dispositivo debe ser listado después de obtener la aprobación y antes de su comercialización en los US. Algunos dispositivos, como los dispositivos de radiación, deben cumplir con otros requisitos, como el número de acceso, antes de poder ser importados a los US.
- Identificación Única de Dispositivos: Todas las clases de dispositivos deben cumplir con las regulaciones de Identificación Única de Dispositivos (UDI) para comercializar los dispositivos en los US.
- Tasas de Establecimiento: El fabricante debe pagar las tasas anuales de establecimiento para mantener activa su inscripción de establecimiento y para seguir comercializando dispositivos en los US. La US FDA ha reducido la estructura de tasas para entidades más pequeñas con un Certificado de Pequeña Empresa activo.
- Auditorías de Calidad: Para los dispositivos que no están exentos de GMP, la US FDA puede inspeccionar el establecimiento de fabricación en cualquier momento para verificar el cumplimiento de las Regulaciones de Sistemas de Calidad (QSR) de acuerdo con el 21 CFR 820.
Flujo del proceso
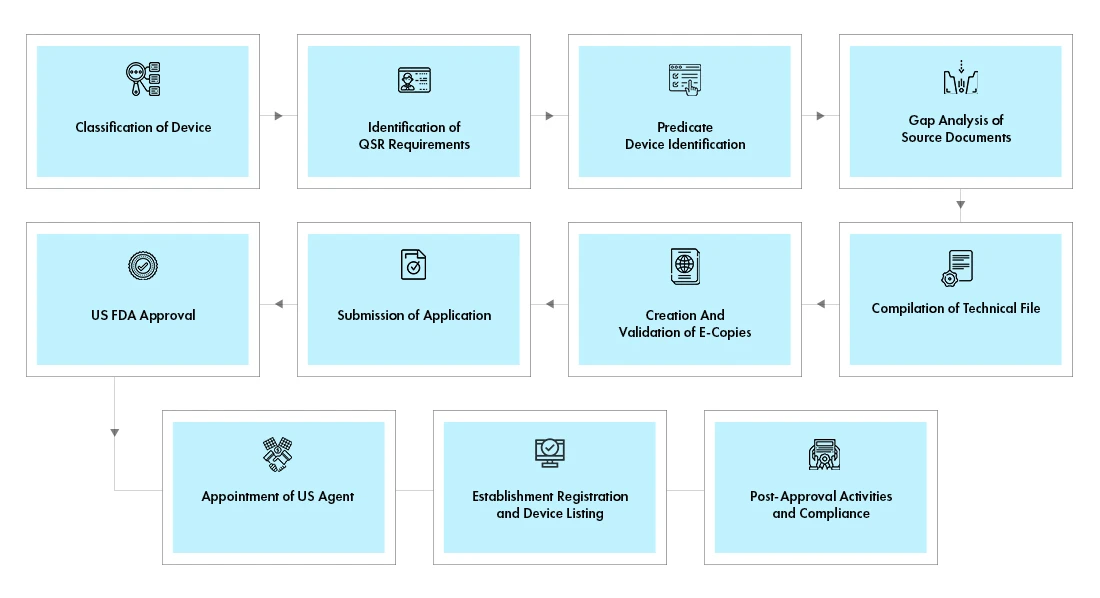
Gestión del ciclo de vida de dispositivos post-aprobación
Freyr apoya a fabricantes extranjeros en la gestión End-to-End del ciclo de vida de Dispositivos Médicos, incluyendo actividades posteriores a la aprobación, tales como:
- Gestión de cambios post-aprobación: modificaciones a las aprobaciones existentes de Dispositivos Médicos, como la adición de nuevas variantes, accesorios; adición de nuevas indicaciones de uso, entre otros.
- Mantenimiento de las aprobaciones y el registro mediante el pago puntual de las tarifas MDUFA a la FDA
- Sirviendo de enlace entre la US FDA y el fabricante.
Freyr tiene un centro de entrega exclusivo en los US con un equipo profesional para proporcionar apoyo normativo a los fabricantes en el mantenimiento de la calidad y seguridad necesarias para la aprobación. Los expertos en inteligencia de Freyr observan atentamente las actualizaciones normativas y mantienen a los clientes informados sobre los pasos a seguir para el cumplimiento del producto con el estándar actual.
Resumen
| Riesgo | Clase de Dispositivo | Auditoría de SGC | Disponibilidad de producto de referencia | Ruta reglamentaria | US Agente | US Plazos de la FDA |
|---|---|---|---|---|---|---|
| Riesgo bajo | Yo | No | NA | Exento | Sí | 1 Mes |
| Riesgo medio | II | Sí (posterior a la aprobación) | Sí | PMN/510(k) | Sí | 9 - 12 Meses |
| Riesgo medio | II | Sí (posterior a la aprobación) | No | Solicitud de Clasificación De Novo | Sí | 18 - 30 Meses |
| Riesgo alto | III | Sí (previo a la aprobación) | NA | PMA | Sí | 18 - 30 Meses |
Servicios de Registro de Dispositivos Médicos de Freyr
Experiencia de Freyr
- Debida diligencia reglamentaria
- Documentación de Dispositivos Médicos
- Apoyo 513(g)
- Registro 510(k)
- Solicitud De Novo de Clasificación
- Registro de PMA
- 21 CFR 820 cumplimiento
- Apoyo para auditoría BIMO
- MDSAP Cumplimiento
- Apoyo en etiquetado
- Apoyo para la publicación y presentación
- Agente de EE. UU.
- Reuniones de Q-Submission
- Reuniones de RFD y Pre-RFD
- Certificación de pequeña empresa
- Registro de establecimientos y listado de dispositivos
- Cumplimiento reglamentario para Dispositivos Médicos de radiación
- Gestión de Cambios Post-aprobación
- Post Market Surveillance
- Cumplimiento UDI
- Consultoría reglamentaria para la resolución de deficiencias