

Resumen Dispositivos Médicos en Europa
La UE, compuesta por 27 member states elReglamento sobre productos sanitarios (MDR) 2017/745yel Reglamento sobre productos sanitarios para diagnóstico in vitro (IVDR) 2017/746, que se han aplicado íntegramente recientemente. Estos reglamentos forman parte integrante del proceso Dispositivos Médicos en Europa y han sustituido a las directivas. Ambos reglamentos incluyen nuevos requisitos adicionales, y estos serán los procedimientos de regulación centralizados que se deberán seguir para comercializar los productos sanitarios en cualquiera de los 27 países. Dispositivos Médicos extranjeros Dispositivos Médicos que no tengan una sede física en Europa deben designar a un representante autorizado europeo (EAR) para que les ayude a cumplir con estos reglamentos.
![]()
Autoridad reguladora: Autoridad nacional competente![]()
Reglamento: Dispositivos Médicos (MDR) 2017/745, Reglamento sobre productos sanitarios para diagnóstico in vitro 2017/746![]()
Vía reglamentaria: Marcado CE seguido de Registro/Notificación![]()
Representante autorizado: Representante europeo autorizado (EAR) para fabricantes de fuera de la UE![]()
Requisito del SGC: ISO 13485:2016![]()
Evaluación de los datos técnicos: Organismo notificado para el marcado CE
Clasificación de los dispositivos
La clasificación del producto es el primer paso para determinar la vía reglamentaria para el producto en cuestión. Existen unas 22 normas de aplicación para los productos sanitarios y se clasifican en
| Clase | Riesgo |
|---|---|
| Clase I | Bajo |
| Clase IIa | Moderado |
| Clase IIb | Moderado a alto |
| Clase III | Alta |
Del mismo modo, en el caso del DIV, se aplican unas 7 normas que se clasifican en las cuatro categorías siguientes
| Clase | Riesgo |
|---|---|
| Clase A | Bajo |
| Clase B | Moderado |
| Clase C | Moderado a alto |
| Clase D | Alta |
Dadas las instrucciones especializadas que existen para las distintas clases, identificar la clase correcta de dispositivo es crucial para determinar la vía reglamentaria.
Representante europeo autorizado (EAR)
Todo fabricante extranjero que pretenda lanzar sus productos en la región de la UE está obligado a designar un Representante Europeo Autorizado (EAR) de conformidad con el artículo 11 del EU MDR y el IVDR de EU MDR .
Dispositivos Médicos
Para comercializar los productos sanitarios en la geografía de la UE, es obligatorio obtener el marcado CE. Los fabricantes deben identificar y designar a los notificados, someterse a la evaluación de conformidad y expedir la certificación CE.
Descifrar la información reglamentaria relativa al registro o notificación de dispositivos a través de un sistema de registro en línea puede resultar complicado sin la ayuda de un experto.
Flujo del proceso
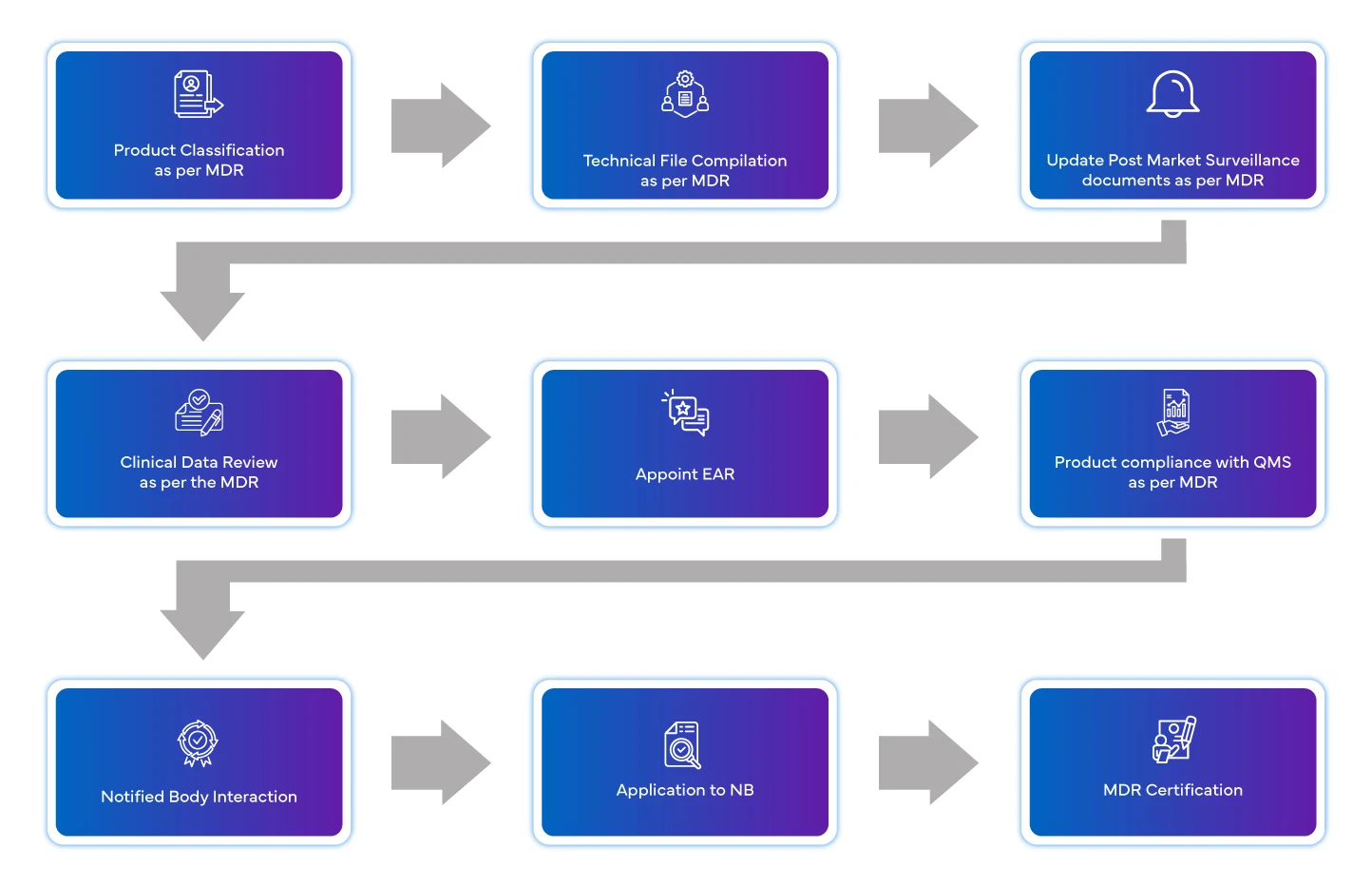
Gestión del ciclo de vida de los dispositivos tras su homologación
La normativa europea sobre productos sanitarios destaca ahora la importancia de los requisitos posteriores a la comercialización. Se exige al fabricante que se dote de un sistema de gestión de la vigilancia. Debe facilitarse información periódica sobre el producto.
Freyr puede ayudarle a elaborar post-market surveillance (PMS) , el informe de vigilancia posterior a la comercialización (PMSR), el informe periódico de seguridad (PSUR) y el seguimiento clínico posterior a la comercialización (PMCF)/seguimiento del rendimiento posterior a la comercialización (PMPF).
Las ayudas Freyr también incluyen actividades como
- Gestión de cambios posteriores a la aprobación: modificaciones de Dispositivos Médicos existentes Dispositivos Médicos , como la incorporación de nuevas variantes o accesorios, la incorporación de nuevas indicaciones de uso, entre otras.
- Mantenimiento de las certificaciones ISO 13485:2016 y CE
- Renovación de licencias
- Enlace entre el organismo notificado y el fabricante
Dispositivos Médicos en Europa
Experiencia Freyr
- Dispositivos Médicos europea de Dispositivos Médicos
- Apoyo del Representante Autorizado Europeo (EAR)
- ISO 14971:2019 Consulta sobre gestión de riesgos
- Cumplimiento de la ISO 13485:2016
- Revisión, compilación y presentación del expediente técnico y de diseño de la CE
- Apoyo a la transición EU MDR
- Apoyo a la transición al IVDR de la UE
- Clinical Evaluation Reports (CER) de productos sanitarios
- Informes de evaluación del funcionamiento (PER) de los productos para diagnóstico in vitro
- Notificación/registro de productos sanitarios a través del sistema de registro en línea
- Informe sobre la estrategia Dispositivos Médicos
- Pruebas de biocompatibilidad, seguridad eléctrica, mecánica y rendimiento
- Apoyo al cumplimiento de las normas de etiquetado
- Apoyo GMP
- Apoyo a la vigilancia posterior a la comercialización
