

Dispositivos Médicos en Indonesia: descripción general
Indonesia puso en marcha la asistencia sanitaria universal para sus ciudadanos en 2014. Esto ha influido enormemente en el crecimiento del mercado de productos sanitarios y ha provocado un aumento de las importaciones de este tipo de productos. Los productos sanitarios en Indonesia están regulados por la Agencia Nacional de Control de Medicamentos y Alimentos (NADFC), dependiente del Ministerio de Salud de Indonesia (MoH). La normativa más reciente en vigor para la importación de productos sanitarios es el Decreto n.º 62, promulgado en 2017. Las empresas extranjeras deben designar a un representante local autorizado en Indonesia para el proceso de Dispositivos Médicos en Indonesia.
![]()
Autoridad reguladora: Agencia Nacional de Control de Medicamentos y Alimentos (NADFC)![]()
Reglamento: N.º 62 / 2017![]()
Representante autorizado: Indonesia Representante local autorizado![]()
Requisito del SGC: ISO 13485:2016![]()
Evaluación de los datos técnicos: NADFC![]()
Validez de la licencia: 5 años![]()
Requisitos de etiquetado: N.º 62 / 2017![]()
Formato de presentación: En línea/Papel![]()
Idioma: Inglés e indonesio
Dispositivos Médicos en Indonesia
La normativa actual clasifica los dispositivos en A, B, C y D en función del riesgo.
| Criterios de riesgo | Clase de dispositivo |
|---|---|
| Riesgo bajo | A |
| Riesgo bajo moderado | B |
| Riesgo moderado - alto | C |
| Alto riesgo | D |
Representante local autorizado en Indonesia
La normativa indonesia exige a los fabricantes que designen a un representante local con licencia de distribuidor. Se puede designar a un distribuidor para que represente al fabricante extranjero en Indonesia. Sin embargo, nombrar a un tercero independiente proporcionaría flexibilidad para cambiar de distribuidor o nombrar a varios distribuidores para una mejor penetración en el mercado.
Dispositivos Médicos en Indonesia
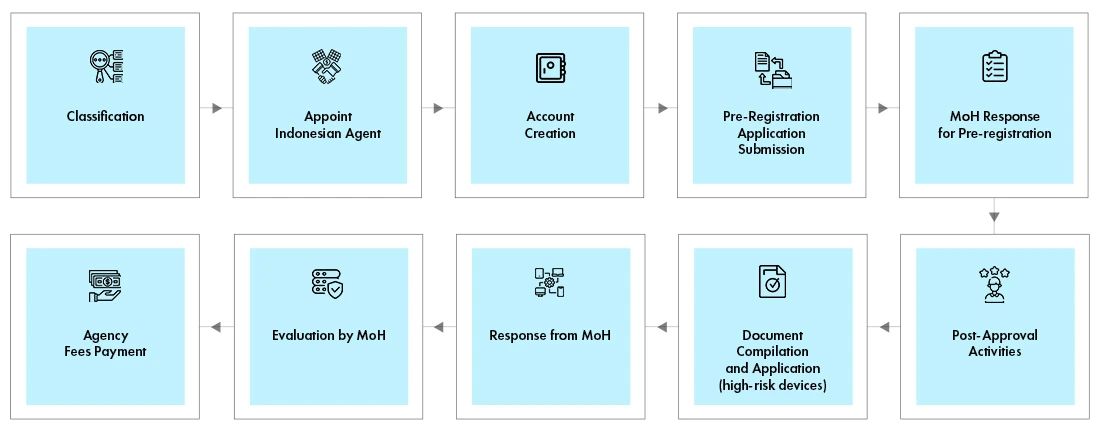
El representante local debe crear una cuenta en el portal en línea. El proceso de registro es el mismo para todas las clases de dispositivos. Sin embargo, los requisitos de documentación varían según la clase de dispositivo. El proceso de registro consta de dos fases
- Proceso de prerregistro
- Proceso de evaluación
El Ministerio de Sanidad verifica la clasificación del dispositivo y determina el coste de la evaluación. El resultado del prerregistro, junto con la factura, se envía por correo electrónico al solicitante. El representante local, en nombre del fabricante, efectuará el pago y cargará el justificante de pago. El Ministerio de Sanidad revisará los documentos y comunicará los resultados por correo electrónico al solicitante. Algunos dispositivos requieren pruebas en el país en un laboratorio acreditado.
Proceso de aprobación reglamentaria
El equipo de expertos de Freyrrealiza un seguimiento de las tendencias y normativas cambiantes y ayuda a las partes interesadas a mantener el cumplimiento normativo durante todo el ciclo de vida del producto. Ofrecemos soluciones normativas para mantener otros aspectos normativos del cumplimiento dentro de presupuestos limitados.
Clase de dispositivo | Clase de riesgo | Plazos del Ministerio de Sanidad para Autorización de comercialización | Plazos del Ministerio de Sanidad para Renovación / Variación | ||
|---|---|---|---|---|---|
| Proceso de clasificación (días) | Proceso de evaluación (días) | Proceso de clasificación (días) | Proceso de evaluación (días) | ||
| Clase A | Riesgo bajo | 7 | 45 | 7 | 45 |
| Clase B | Riesgo bajo moderado | 7 | 90 | 7 | 45 |
| Clase C | Riesgo moderado - alto | 7 | 100 | 7 | 45 |
| Clase D | Alto riesgo | 7 | 120 | 7 | 45 |
Experiencia Freyr
- Diligencia debida reglamentaria
- Registro de dispositivos
- Pruebas en el país
- Licencias para distribuidores
- Legalización y notarización
- Representante legal
- Labelling
- Apoyo a la traducción
- Identificación y cualificación de distribuidores
- Servicios de vigilancia posterior a la comercialización
- Gestión de cambios tras la aprobación
- Servicios de renovación y transferencia de licencias
- Servicios de presentación y enlace
