SaMD en EE. US descripción general
Respuestas en esta página
- ¿Es mi software un SaMD?
- ¿Qué clase de riesgo de SaMD requiere un 510(k)?
- Registro y cumplimiento de SaMD : el proceso 510(k)
- ¿Durante cuánto tiempo es válida la autorización?
¿Es mi software un SaMD?
Según el Foro Internacional Dispositivos Médicos (IMDRF), SaMD :
- Programas informáticos destinados a uno o varios usos médicos.
- Realiza estas funciones sin formar parte de un Dispositivos Médicos de hardware.
¿Qué clase de riesgo de SaMD requiere un 510(k)?
Determinar la SaMD de su software es un paso importante en el proceso de registro. Una vez que su software ha sido clasificado como SaMD, es esencial comprender la vía reglamentaria que requiere para acceder al mercado US. SaMD clasifican normalmente en diferentes clases, en función de sus niveles de riesgo. SaMD de clase II SaMD considera de riesgo moderado, requiere una autorización 510(k) y se basa en demostrar una equivalencia sustancial con el dispositivo predicado comercializado legalmente. El proceso de autorización garantiza que su SaMD sustancialmente equivalente a los dispositivos existentes, lo que ayuda a garantizar su seguridad y eficacia antes de su comercialización.
Registro y cumplimiento de SaMD : el proceso 510(k)
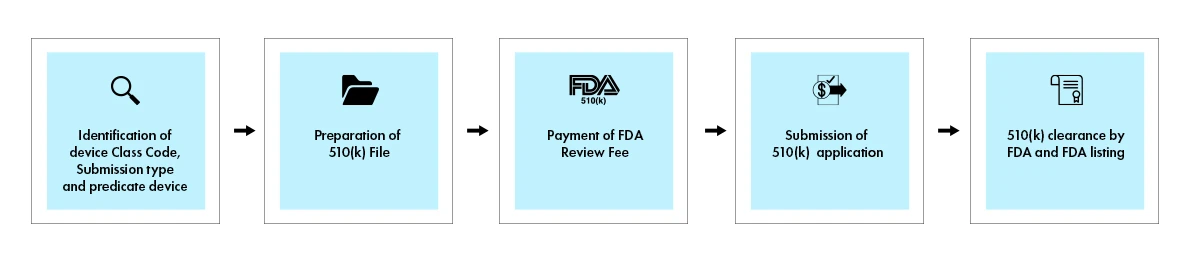
El proceso 510(k) implica una presentación exhaustiva que demuestre la equivalencia sustancial con un dispositivo predicado comercializado legalmente. Cuando se toma una decisión, la Administración US y Medicamentos US (FDA) envía una carta de decisión al solicitante por correo electrónico. Una solicitud 510(k) que recibe una carta de decisión de equivalencia sustancial se considera «aprobada». A continuación, se incluye en la base de datos 510(k), con el resumen 510(k) adjunto. La siguiente figura ofrece una visión general de los pasos clave del proceso 510(k).
¿Durante cuánto tiempo es válida la autorización?
Una autorización 510(k) sigue siendo válida hasta que se produzcan cambios significativos en el dispositivo o en la normativa aplicable. Sin embargo, es importante señalar que laFDA US FDA solicitar informes periódicos o información adicional para garantizar el cumplimiento y la seguridad continuos.
En conclusión, el registro SaMD requiere un conocimiento profundo de la clasificación SaMD , el cumplimiento SaMD y los procesos regulatorios. Recurrir a los servicios de consultoría de SaMD puede proporcionar orientación experta para navegar por las complejidades y garantizar un resultado de registro exitoso.
SaMD en EE. US
- EstrategiaFDA integralFDA US para SaMD.
- Clasificación SaMD .
- Identificación del dispositivo de predicado.
- Establecer la equivalencia sustancial con el dispositivo predicado.
- Análisis de deficiencias paraFDA de la normativa deFDA US .
- Recopilación del expediente técnico 510(k), de acuerdo con la Guía de presentación previa a la comercialización de softwareFDA US
- Creación de la plantilla eCopy/eSTAR.
- Validación y presentación de la plantilla eCopy/eSTAR.
- Servicios de enlace para la aprobación de dispositivos.
- Apoyo a la respuesta RTA y a las deficiencias AINN.
- Servicios de consulta para subsanar deficiencias.
- Registro de establecimiento ante laFDA US
- Listado de dispositivos y mantenimiento de la base de datos FURLS.
- Servicios de representante legal (RL).

- Amplia experiencia con diversos registros 510(k).
- Experiencia en la recopilación de información para la notificación previa a la comercialización (510[k])FDA US , de conformidad con los requisitos de dicha notificación.
- Soporte adicional para la gestión de consultas 510(k).
- Presentación puntual de los resultados.
- Al día con las nuevas enmiendasFDA US sobre SaMD.





