Declaración de trabajo (SOW) para Dispositivos Médicos activos Dispositivos Médicos Dispositivos Médicos no activos. Resumen de la presentación Dispositivos Médicos (K).
En Freyr, nuestro equipo de expertos recopila y sintetiza diligentemente la información más reciente y esencial para sus solicitudes 510(K), tanto para dispositivos médicos activos como no activos. Esto le garantiza que dispondrá de los conocimientos necesarios para desenvolverse con confianza en el marco normativo. Desde aclarar las diferencias entre los dispositivos activos y no activos hasta profundizar en las complejidades de la presentación 510(K), hemos recopilado un amplio repositorio de recursos para su referencia principal. Emprenda un viaje hacia el dominio de la presentación Dispositivos Médicos (K) Dispositivos Médicos activos y la presentación 510(K) de dispositivos médicos no activos con nuestra guía completa.
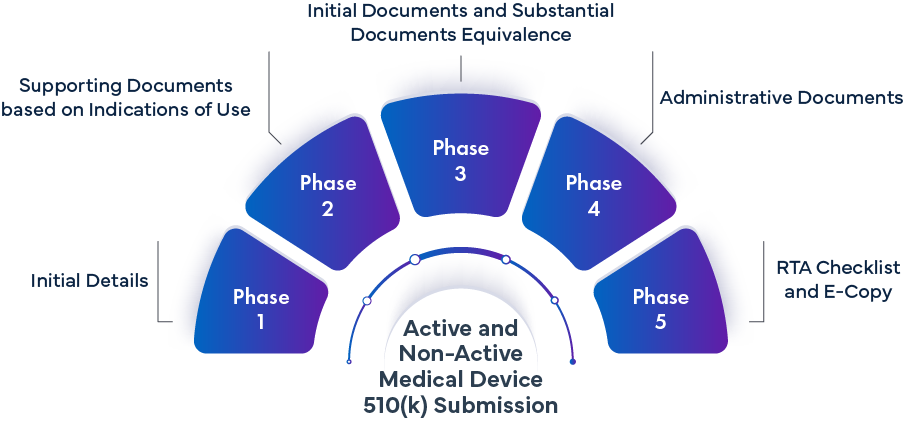
Fase -1 Detalles iniciales | ||
|---|---|---|
Requisitos | Alcance de la solicitud 510(k) | Ámbito de Freyr |
| Uso previsto |
|
|
| Declaración de Indicaciones de Uso (Form3881) |
|
|
| Descripción del dispositivo |
|
|
| Normas y orientaciones |
|
|
| Dispositivo de predicado |
|
|
| 510(K) Resumen |
|
|
Fase 2: Documentación justificativa basada en las indicaciones de uso | |||
|---|---|---|---|
Requisitos de los documentos | Alcance de la solicitud 510(k) | Ámbito de Freyr | |
| 2.1 | Dibujo del dispositivo | Presentar el archivo de dibujo del dispositivo para garantizar una representación exacta del diseño del dispositivo. | Inicie una solicitud formal de dibujo de un dispositivo activo. Revisar minuciosamente y documentar meticulosamente la información necesaria para la presentación del 510(k). |
| 2.2 | Diseño y desarrollo del dispositivo | Presentar el expediente de diseño y desarrollo del dispositivo Activo, que incluya toda la información y documentación pertinentes. | Presente una solicitud para el diseño y desarrollo de un dispositivo activo. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.3 | Ficha de datos de seguridad | Proporcione las fichas de datos de seguridad de los materiales (MSDS) para los componentes esenciales del dispositivo activo, asegurando que se incluya información completa sobre su seguridad y composición. | Envíe una solicitud de hoja de datos de seguridad de los componentes cruciales del dispositivo activo. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.4 | Diagrama de flujo de fabricación | Suministrar un diagrama de flujo de fabricación en el que se detalle el proceso de producción del dispositivo activo y se ofrezca una representación visual de las etapas de fabricación y su secuencia. | Envíe una solicitud para obtener la ficha de datos de seguridad (MSDS) de los componentes esenciales del dispositivo activo. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.5 | Descripción del dispositivo | Proporcione detalles completos que incluyan: o Una visión general del dispositivo o Funciones y modos de funcionamiento o Diagramas de bloques o Fotografías, cables y accesorios pertinentes o Interoperabilidad de dispositivos. o Descripción de la fuente de alimentaciónTop of Form | Envíe una solicitud de información detallada sobre el dispositivo. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.6 | Labelling propuesto | Proporcione las Instructions for Use (IFU), el manual del usuario y cualquier material promocional asociado al dispositivo activo. | Envíe una solicitud para obtener las Instructions for Use (IFU), el manual del usuario y cualquier material promocional, si está disponible. Revise la IFU, el manual de usuario y el material promocional proporcionado por el solicitante. Documente las instrucciones de uso, el manual del usuario y el material promocional a efectos de la presentación de la solicitud 510(k). |
| 2.7 | Embalaje y transporte | Proporcionar los planes de estudio y los informes para la validación del envasado y el transporte. | Presentar una solicitud para el plan de estudio y los informes relativos a la Validación de Embalaje y Transporte. Revisar los planes e informes de estudio para la Validación de Embalaje y Transporte y aportar las correcciones o comentarios necesarios. |
| 2.8 | Esterilización (si procede) | Proporcionar los planes de estudio e informes para la Validación de la Esterilización. | Presentar una solicitud para el plan de estudio y los informes de validación de la esterilización. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.9 | Pruebas de rendimiento _ Banco | Iniciar una solicitud formal para los planes e informes del estudio del banco de pruebas de rendimiento, describiendo los requisitos y objetivos específicos que deben abordarse. | Presentar una solicitud de los planos e informes del estudio en banco del dispositivo activo para las pruebas de rendimiento. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
Compatibilidad electromagnética y seguridad eléctrica Documentación de apoyo | |||
| 2.10 | Características de los dispositivos relacionados con la CEM y entornos de uso previstos | Proporcione detalles sobre las características de los dispositivos relacionados con la CEM y los entornos de uso previstos, incluidos: o Una visión general del dispositivo. o Funciones y modos de funcionamiento. o Diagramas de bloques. o Fotografías, cables y accesorios pertinentes. o Interoperabilidad de dispositivos. o Descripción de la fuente de alimentación, incluida la viabilidad de utilizar los Dispositivos Médicos con alimentación interna Dispositivos Médicos se cargan. o Entornos en los que Dispositivos Médicos prevé utilizar los Dispositivos Médicos . o Descripción de cualquier tecnología inalámbrica (si procede) para consideraciones adicionales relativas a los dispositivos médicos inalámbricos. o Descripción de cualquier emisor de radiofrecuencia interno en los Dispositivos Médicos pudiera causar perturbaciones electromagnéticas. o Abordar los emisores electromagnéticos (EM) comunes, así como los emisores médicos únicos.
| Presente una solicitud de información sobre las características de los dispositivos relacionados con la CEM y los entornos de uso previstos. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.11 | Evaluación de riesgos | Suministrar un Plan de Gestión de Riesgos que incluya una evaluación de riesgos que muestre la mitigación efectiva de los mismos, junto con un informe exhaustivo de gestión de riesgos que abarque todos los elementos de riesgo. Entregar el documento revisado con las correcciones y mejoras sugeridas | Presente una solicitud para el Expediente de Gestión de Riesgos y solicite la documentación del Plan y el Informe de Gestión de Riesgos, incluida la identificación de los peligros de riesgo, la evaluación de riesgos y la demostración de la mitigación de riesgos adecuada. El Informe de gestión de riesgos debe abarcar todos los elementos de riesgo, preferiblemente con secciones separadas para mayor claridad. Proporcionar una plantilla del Plan de Gestión de Riesgos y del Informe de Gestión de Riesgos que abarque todos los riesgos relacionados con el dispositivo a petición del solicitante. Revisar los datos del Expediente de Gestión de Riesgos, incluidos el Plan y el Informe compartidos por el solicitante, y aportar sugerencias sobre las correcciones necesarias para garantizar una documentación completa para la presentación del 510(k). Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.12 | Norma de consenso | Proporcione confirmación de las normas consensuadas pertinentes y una explicación de cualquier desviación de las normas FDA. | Presente una solicitud de las normas consensuadas aplicables relacionadas con la CEM y la seguridad eléctrica para el dispositivo Activo. Documentar las normas de consenso confirmadas para el dispositivo activo a efectos de la presentación del 510(k). |
| 2.13 | Rendimiento esencial e inmunidad Criterios de aprobado/suspenso | Presente el plan de estudio y los informes de las pruebas de rendimiento esencial e inmunidad realizadas en el dispositivo activo, cumpliendo con las normas FDA. | Envíe una solicitud para obtener el plan de estudio y los informes de las pruebas de rendimiento esencial y de inmunidad realizadas en el dispositivo activo, de conformidad con las normas FDA. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.14 | Dispositivos Médicos y funciones probadas | Proporcione la Dispositivos Médicos y las funciones probadas de Dispositivos Médicos para el dispositivo activo, incluyendo los siguientes detalles: o Proporcione una descripción exhaustiva de los Dispositivos Médicos prueba, incluyendo información detallada sobre su configuración, funciones, modos y los ajustes específicos que se han probado. o La descripción del dispositivo sometido a prueba debe incluir el Dispositivos Médicos , el número de modelo e indicar si el dispositivo es el Dispositivos Médicos final listo para la producción que se está evaluando Dispositivos Médicos . | Envíe una solicitud para las funciones de configuración y prueba de los Dispositivos Médicos activos. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
| 2.15 | Resultados de las pruebas CEM | Proporcione el plan de estudio y el informe de pruebas de compatibilidad electromagnética (EMC) de acuerdo con la norma consensuada FDA recomendada para el dispositivo activo. | Iniciar una solicitud formal para el plan de estudio y el informe de pruebas de compatibilidad electromagnética, en consonancia con la norma consensuada FDA recomendada para dispositivos activos. Revise a fondo y documente meticulosamente toda la información necesaria para preparar la presentación del 510(k). |
Fase 3 - Documentos iniciales y documentos de equivalencia sustancial | |||
|---|---|---|---|
Requisitos de los documentos | Alcance de la solicitud 510(k) | Ámbito de Freyr | |
| 3.1 | Hoja de presentación de la revisión previa a la comercialización del CDRH (FDA 3514FDA ) | - | Rellene el FDA 3514 FDA utilizando los datos proporcionados por el solicitante. |
| 3.2 | Resumen y certificación de la Clase III | - | Este paso no es necesario si no se requieren estudios clínicos |
| 3.3 | Certificación financiera o declaración informativa | - | Este paso no es necesario si no se requieren estudios clínicos |
| 3.4 | Resumen ejecutivo | - | Elabore una plantilla y prepare meticulosamente el documento. Justifique las discrepancias observadas entre el dispositivo propuesto y el dispositivo de referencia. Estudio comparativo entre el dispositivo propuesto y el dispositivo predicado se elige, crear una plantilla y preparar el documento correspondiente. |
| 3.5 | Debate sobre la equivalencia sustancial | - | Elabore una plantilla y prepare meticulosamente el documento. Estudio comparativo entre el dispositivo propuesto y el dispositivo predicado se elige, crear una plantilla y preparar el documento correspondiente. |
Fase 4 - Documentos administrativos | |||
|---|---|---|---|
Requisitos de los documentos | Alcance de la solicitud 510(k) | Ámbito de Freyr | |
| 4.1 | 510(k) Carta de presentación | Firme el documento impreso en papel membretado de la empresa y envíe una copia impresa por mensajería a la US Proporcionar una copia digital de la carta de presentación firmada del 510(k) para su inclusión en la documentación del 510(k). | Prepare una plantilla completa que incluya todos los detalles necesarios para la carta de presentación y entréguela al solicitante. Indique al solicitante que utilice su membrete oficial y asegúrese de que la carta de presentación esté firmada por una persona autorizada. |
| 4.2 | Declaración de veracidad y exactitud | Asegúrese de que el documento está firmado por la persona de contacto designada en la empresa y de que se facilita en consecuencia. | Elabore una plantilla completa con todo el contenido necesario que debe incluirse en el documento de presentación. |
| 4.3 | Declaraciones de conformidad e informe de síntesis | Asegúrese de que el documento está firmado por la persona de contacto designada en la empresa y de que se facilita en consecuencia. | Elabore una plantilla completa para enumerar y preparar sistemáticamente los documentos necesarios. |
| 4.4 | MDFUSC (FDA 3601FDA ) | Envíe el pago requerido a la FDA de la presentación formal del expediente 510(k). | Genere una portada de tarifa de usuario y un número de identificación personal (PIN) único específicamente para Dispositivos Médicos . |
Fase 5 - Lista de control RTA y copia electrónica | |||
|---|---|---|---|
Requisitos de los documentos | Alcance de la solicitud 510(k) | Ámbito de Freyr | |
| 5.1 | Lista de control RTA | Aprobación de la lista de verificación RTA (Ready to Accept), que indica que se han cumplido satisfactoriamente todos los requisitos. | Elabore una plantilla de lista de comprobación de ACR adaptada al tipo específico de presentación. Complete la lista de verificación rellenando meticulosamente todos los campos obligatorios y asegurándose de que los documentos mencionados se envíen debidamente a la FDA se compartan con el solicitante. |
| 5.2 | E-Copy | Aprobación de la documentación contenida en la carpeta de presentación final, en la que conste que cumple todos los requisitos y normas necesarios. | Organice las secciones de la carpeta de presentación de acuerdo con FDA y compártalas sin demora con el solicitante. Convierta la carpeta de presentación en una copia electrónica para facilitar el acceso y la revisión. Envíe la copia electrónica de la presentación al US designado US |
Dispositivos Médicos
- EstrategiaFDA integral deFDA US
- Identificación del dispositivo predicado
- Establecer la equivalencia sustancial con el dispositivo predicado
- Análisis de deficiencias paraFDA de la normativa deFDA US
- Recopilación de 21 secciones del Expediente Técnico 510(k)
- Publicación y creación de eCopy
- Validación y presentación de eCopy
- Servicios de enlace para la aprobación de dispositivos
- Respuesta y deficiencias de RTA
- Servicios de consulta para subsanar deficiencias
- Listado de dispositivos y mantenimiento de la base de datos FURLS

- Ha gestionado numerosos registros 510(k) de diversas categorías de dispositivos
- Equipo experto en la recopilación de información para la notificaciónFDA (510(k)) según los requisitos deFDA US
- Apoyo adicional para gestionar las consultas 510(k)
- Asesoramiento sobre el tipo adecuado de 510(k) según los requisitos de presentación deFDA (k) US para el dispositivo.
- Presentación puntual de los resultados
- Actualizado con lasFDA enmiendas deFDA US
