

Resumen Dispositivos Médicos en Taiwán
Taiwán tiene una demanda creciente de dispositivos médicos. La Administración de Alimentos y Medicamentos de Taiwán (TFDA), dependiente del Ministerio de Salud y Bienestar Social (MOHW), regula los dispositivos médicos a través de la Ley de Asuntos Farmacéuticos (PAA). Los fabricantes extranjeros que no tengan una oficina física en Taiwán deben contar con un representante en Taiwán como requisito previo para el proceso Dispositivos Médicos en Taiwán.
![]()
Autoridad reguladora: Administración de Alimentos y Medicamentos de Taiwán![]()
Reglamento: Ley de asuntos farmacéuticos (PAA) y Reglamento para el registro de productos sanitarios![]()
Representante autorizado: Se requiere la representación de un agente en Taiwán![]()
Requisito del SGC: Documentación del sistema de calidad (QSD) ISO 13485![]()
Evaluación de los datos técnicosa: División de Productos Sanitarios y Cosméticos![]()
Validez de la licencia: QSD - 3 años; Registro del producto - 5 años![]()
Requisitos de etiquetado: Artículo 75 de la Ley de asuntos farmacéuticos![]()
Formato de presentación: Papel![]()
Idioma: Inglés y chino
Dispositivos Médicos de Taiwán
La TFDA clasifica los productos sanitarios en 3 clases en función del riesgo: Clase I para los de bajo riesgo, Clase II para los de riesgo moderado y Clase III para los de alto riesgo. La necesidad de un dispositivo de referencia supone un reto para la introducción en el mercado de nuevos dispositivos. Otra de las complejidades es el aumento del tiempo de procedimiento para los productos de las clases II y III que necesitan documentación del sistema de calidad. Todos los productos sanitarios importados deben obtener un certificado de registro de la TFDA.
| Clase de dispositivo | Riesgo |
|---|---|
| Clase I | Riesgo bajo |
| Clase II | Riesgo moderado |
| Clase III | Alto riesgo |
Representación de agentes en Taiwán
Los fabricantes extranjeros sin oficina física en Taiwán deben nombrar a un Agente de Taiwán como requisito previo para comercializar dispositivos en Taiwán. El nombramiento de una organización externa como Agente de Taiwán en lugar de distribuidor ofrece flexibilidad para explorar múltiples distribuidores para una mejor penetración en el mercado. El Agente de Taiwán debe tener una entidad legal establecida en Taiwán, certificada con una Licencia de Ventas Farmacéuticas.
Dispositivos Médicos en Taiwán
Antes de que un Dispositivos Médicos venderse en Taiwán, además del Dispositivos Médicos , es necesario registrar la documentación del sistema de calidad (QSD) de la planta de fabricación. Solo se exime del registro QSD a los dispositivos médicos de clase I (no estériles). La licencia QSD (que se obtiene tras la aprobación del registro QSD) en Taiwán es similar a las buenas prácticas de fabricación (GMP) para dispositivos médicos.
La TFDA anunció que, a partir del 1 de junio de 2022, los titulares de licencias de dispositivos médicos de clase III deberán cargar el UDI y la información correspondiente del producto en la base de datos UDI (UDID). Dispositivos Médicos también deberán incluir el UDI en la etiqueta del producto. Además, a partir del 1 de junio de 2023, los dispositivos médicos de clase II deberán cumplir con las normativas pertinentes del UDI.
Flujo del proceso
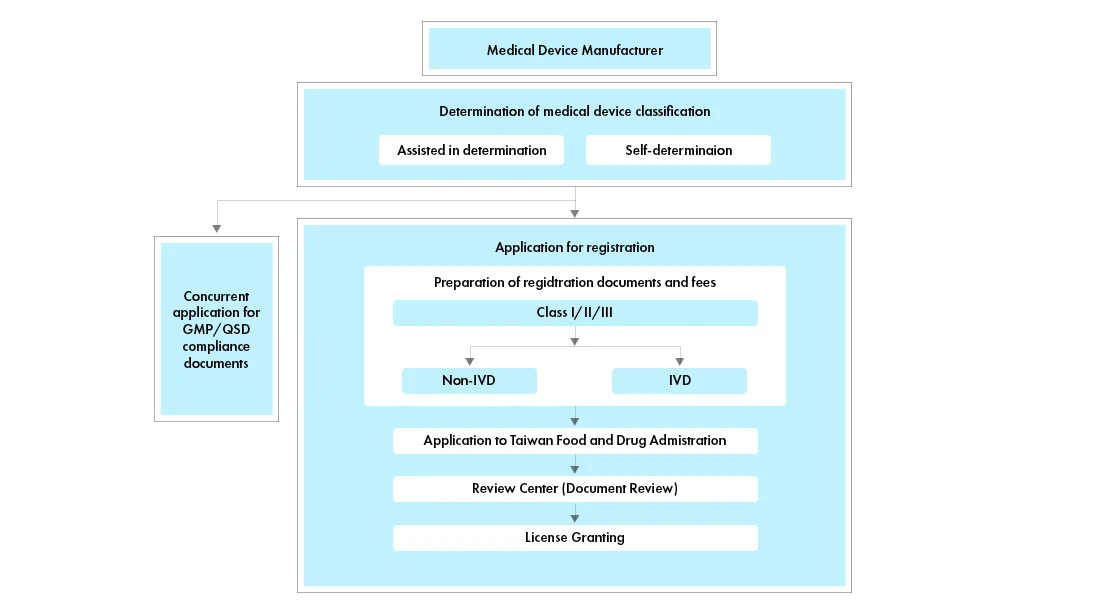
Gestión del ciclo Dispositivos Médicos tras su aprobación
Freyr presta apoyo a los fabricantes extranjeros en la gestión end-to-end Dispositivos Médicos , incluidas las actividades posteriores a la autorización, tales como:
- Gestión de cambios posteriores a la aprobación: modificaciones de Dispositivos Médicos existentes Dispositivos Médicos , como la incorporación de nuevas variantes, accesorios o indicaciones de uso, entre otras.
- Mantenimiento de las autorizaciones y el registro mediante el pago puntual de las tasas administrativas y de registro.
- Renovación de licencias
- Enlace entre la TFDA y el fabricante
- Gestión de la importación
Freyr se especializa en atender las necesidades regulatorias de Dispositivos Médicos en Taiwán. Con una amplia red, Freyr ayuda en la designación de un agente local fiable cuya presencia es de suma importancia a lo largo de la vigilancia posterior a la comercialización. Nuestros expertos también ayudan en la selección del dispositivo predicado adecuado y las aprobaciones existentes de otros mercados para apoyar la entrada en el mercado de nuevos dispositivos.
Resumen
| Clase de dispositivo | Riesgo / Criterios de clasificación | SGC | Registro de productos |
|---|---|---|---|
| Clase I | Riesgo bajo | Exentos (productos de clase I no estériles) | Sí |
| Clase II | Riesgo moderado | QSD | Sí |
| Clase III | Alto riesgo | QSD | Sí |
Experiencia Freyr
- Diligencia debida reglamentaria
- Clasificación oficial
- Homologaciones QSD
- Registro de dispositivos
- Representante legal
- Soporte de etiquetado
- Apoyo a la traducción
- Identificación y cualificación de distribuidores
- Vigilancia posterior a la comercialización
- Gestión de cambios tras la aprobación
- Renovación y transferencia de licencias
- Presentación y enlace
