USFDA Dispositivos Médicos : resumen de la aprobaciónDispositivos Médicos
El proceso de aprobación USFDA (PMA) USFDA FDA US es una de las vías de registro de dispositivos que ofrece laFDA US , diseñada principalmente para los dispositivos médicos FDA III FDA . El proceso de aprobación FDA para los dispositivos de clase III implica meticulosas evaluaciones científicas y normativas para evaluar la seguridad y la eficacia Dispositivos Médicos, lo que garantiza que se cumplan los más altos estándares antes de la autorización de comercialización.
Concierte una reunión con nuestros expertos en aprobación previa a la comercialización
¿Quién debe presentar una solicitud de aprobaciónDispositivos Médicos (PMA)Dispositivos Médicos USFDA ?
Los fabricantes de productos deben presentar una solicitud de PMA si el producto:
- Es novedoso.
- Pertenece a una clase de alto riesgo.
- No se encuentra en la base de datos de clasificación de productos.
- No es sustancialmente equivalente (NSE) a los dispositivos de clase I, II o III.
Obtenga asesoramiento experto sobre su solicitud de aprobación previa a la comercialización
¿Cuál es la diferencia entre las solicitudes 510(k), PMA y De-Novo?
Aprobación previa a la comercialización
- Producto perteneciente a la clase III que sustenta la vida humana o que presenta un riesgo potencial e irrazonable de enfermedad o lesión.
- El proceso de aprobación PMA FDA requiere ensayos clínicos.
- Requiere inspección in situ antes de emitir la aprobación PMA.
- 180 días naturales
Clasificación De-Novo
- Dispositivos nuevos de las Clases I y II que no tienen un dispositivo predicado válido.
- Requiere datos de estudios clínicos.
- Sin auditoría in situ antes de la aprobación De-Novo.
- 150 días naturales.
Registro 510(k)
- Dispositivos de clase III FDA que tengan una equivalencia sustancial con el dispositivo predicado.
- No requiere pruebas en humanos.
- Sin auditoría in situ antes de la autorización 510(k).
- 90 días naturales.
¿Cuáles son los distintos métodos de solicitud de aprobación previa a la comercialización de FDA ?
Los fabricantes pueden optar por cualquiera de los siguientes cuatro (04) métodos de solicitud de PMA que mejor se adapten a su dispositivo:
- PMA tradicional
- PMA modular
- Protocolo de desarrollo de productos
- Exención de dispositivos humanitarios
¿Cuáles son los requisitos de datos para la aprobación Dispositivos Médicos ?
De acuerdo con la norma 21 CFR parte 814, los solicitantes deben presentar ante laFDA US un formulario de solicitud del CDRH debidamente cumplimentado, los compromisos requeridos y un expediente técnico PMA bien redactado. El expediente técnico deberá incluir los datos no clínicos y clínicos.
Datos no clínicos - Consisten en datos sobre microbiología, toxicología, inmunología, biocompatibilidad, estrés, desgaste, vida útil y otras pruebas de laboratorio o con animales.
Datos clínicos - Consisten en datos sobre protocolos de estudio, datos de seguridad y eficacia, reacciones adversas y complicaciones, fallos y sustituciones de dispositivos, información sobre pacientes, quejas de pacientes, tabulaciones de datos de todos los sujetos individuales, resultados de análisis estadísticos y cualquier otra información de las investigaciones clínicas.
¿En qué consiste el proceso de solicitud de la PMA?
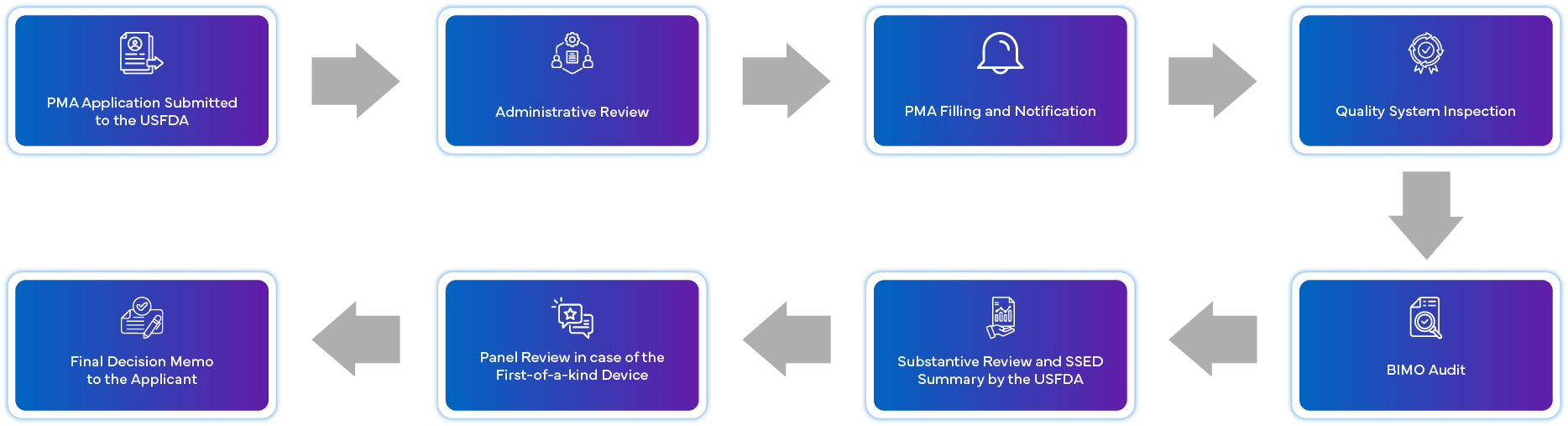
¿Cuáles son los requisitos de cumplimiento posteriores a la aprobación de la PMA?
Los productos aprobados por la vía de la PMA deberán cumplir los requisitos postcomercialización establecidos por USFDA. El producto debe cumplir lo siguiente:
- Requisitos posteriores a la aprobación impuestos en la orden de aprobación PMA FDA .
- Gestión de los cambios posteriores a la aprobaciónmediante la presentación oportuna de los suplementos pertinentes de la PMA.
- Presentación de informes posteriores a la homologación (anuales)
- Normativa 21 CFR 803 para Dispositivos Médicos (MDR)
- Estudios de vigilancia postcomercializaciónexigidos por USFDA en las órdenes de aprobación PMA.
¿Cuáles son las tasas de USFDA para la revisión de la solicitud de PMA?
Las tasas de usuario del MDUFA para el PMA original y los suplementos son las siguientes-.
| Tipo de aplicación | Tasas para el ejercicio 2023 (del supsupde 2022 al sup de septiembre | |
|---|---|---|
| Tasa estándar | Tasa para pequeñas empresas | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Suplemento Panel-Track | $353,238 | $88,309 |
| Suplemento de 180 días | $66,232 | $16,558 |
| Tasa anual por la presentación de informes periódicos sobre un producto de la clase III (PMA, PDP y PMR) | $15,454 | $3,864 |
| Aviso de 30 días | $7,065 | $3,532 |
| Suplemento en tiempo real | $30,908 | $7,727 |
Con experiencia en la tramitación de solicitudes PMA, Freyr puede ayudar a identificar y recopilar la información y asistir en la preparación y revisión de la solicitud.
USFDA Dispositivos Médicos Experiencia y ventajas en la aprobaciónDispositivos Médicos
- Diligencia debida reglamentaria
- Cumplimiento de la inspección del sistema de calidad
- Cumplimiento de la auditoría BIMO
- Recopilación de fichas técnicas PMA
- Publicación y creación de eCopy
- Validación y presentación del eCopy
- Respuesta y deficiencias de RTA
- Servicios de enlace hasta la aprobación previa a la comercialización de FDA
- Consulta por deficiencias
- Listado de dispositivos y registro de establecimientos
- Gestión de suplementos de PMA y avisos de 30 días
- Presentación de informes periódicos anuales
- Auditorías simuladas y 21 CFR 820 Formación

- Experiencia en la tramitación de numerosas solicitudes PMA FDA para diversas categorías de dispositivos.
- Equipo de expertos para la solicitud de aprobación previa a la comercialización de FDA conforme a los requisitos reglamentarios
- Apoyo adicional para atender las consultas relacionadas con la PMA.
- Presentación puntual de los resultados
- Al día con lasFDA enmiendas deFDA US
