2 min de lectura
Shonin (Aprobación previa a la comercialización) es la vía reglamentaria para el registro de Dispositivos Médicos en Japón. La vía Shonin es principalmente para el registro de Dispositivos Médicos de Clase II y III para los que no se dispone de los estándares de clasificación de la PMDA. También para los Dispositivos Médicos de Clase IV de alto riesgo, los fabricantes deben presentar la solicitud Shonin. La PMDA es responsable de la revisión y aprobación de la solicitud Shonin.
¿Cuáles son las otras vías de registro de dispositivos en Japón?
Además de Shonin, las vías Todokede y Ninsho también se utilizan para aprobaciones de Dispositivos Médicos en Japón. Los fabricantes de Dispositivos Médicos pueden elegir una de ellas según la clase de riesgo del dispositivo y la disponibilidad de dispositivos precedentes en Japón. El fabricante deberá identificar la clasificación del dispositivo e investigar la disponibilidad de la Norma Industrial Japonesa (JIS) antes de determinar la vía de registro aplicable.
- Todokede (Presentación previa a la comercialización) – Es aplicable a dispositivos de Clase I y requiere que los fabricantes presenten una notificación previa a la comercialización a la PMDA para su aprobación.
- Ninsho (Certificación Precomercialización) - Es aplicable a Dispositivos Médicos genéricos de Clase II y III que cumplen con estándares de certificación (Normas JIS). El Organismo de Certificación registrado (RCB) es responsable de la revisión y aprobación de la solicitud.
¿Cuáles son los requisitos previos para el registro Shonin?
Los fabricantes que registran sus dispositivos a través de la vía Shonin deben planificar meticulosamente las presentaciones. Deben asegurarse de lo siguiente:
- Presentación de datos generales del dispositivo, como la categoría del Dispositivo Médico, el uso previsto, los datos de análisis de riesgo de eficacia, los datos clínicos, etc.
- Proporcionar Resumen de la Documentación Técnica (STED).
- Provisión de documentos solo en idioma japonés.
- Los fabricantes extranjeros deben designar obligatoriamente un Titular de la Autorización de Comercialización (MAH) o un Titular Designado de la Autorización de Comercialización (DMAH)
- Los fabricantes extranjeros deben obtener el certificado de Registro de Fabricantes Extranjeros (FMR) para sus establecimientos de fabricación.
¿Cuáles son los requisitos de QMS para el registro de dispositivos bajo la vía Shonin?
Los fabricantes deben cumplir con todos los requisitos del QMS definidos en la Ordenanza 169. El patrocinador o DMAH o MAH debe solicitar a la PMDA. La PMDA realiza una inspección detallada del QMS de la instalación del fabricante y emite el certificado tras la implementación satisfactoria del QMS.
¿Cuál es el proceso de registro para la aprobación de dispositivos bajo la vía Shonin?
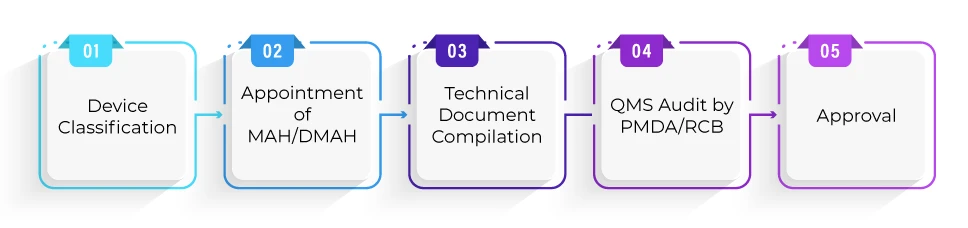
¿Cuál es el plazo promedio necesario para la aprobación de dispositivos según la vía Shonin?
La PMDA generalmente requiere 12 meses para la evaluación técnica a partir de la fecha de recepción de la solicitud Shonin. El fabricante debe considerar el tiempo necesario para preparar los documentos de presentación o realizar estudios clínicos en sus cronogramas de proyecto.
¿Existe algún plazo de caducidad para el registro de dispositivos bajo la vía Shonin?
El registro de Dispositivos Médicos no caduca, pero el patrocinador debe renovar los certificados del SGC cada cinco (05) años.
Japón es un mercado lucrativo, pero intrínsecamente conlleva complejidades regulatorias y barreras lingüísticas. Los fabricantes deben considerar estos factores y planificar proactivamente su estrategia de comercialización (GTM) para Japón. Los fabricantes de dispositivos médicos y de IVD pueden optar por externalizar todos los matices regulatorios a un socio regulatorio fiable y utilizar los recursos para centrarse en otros componentes esenciales.
Para saber más sobre la aprobación de Dispositivos Médicos Shonin en Japón o cualquier otra regulación de PMDA Japón, contacte hoy mismo con los expertos reglamentarios de Freyr.