2 min de lectura
El objetivo principalFDAUS es examinar constantemente y salvar la brecha entre los procesos normativos para la importación y venta ininterrumpidas de dispositivos médicos nuevos y de alta calidad en el US .FDA 1998, laFDA US publicó un programa denominado «El nuevo paradigma 510(k): enfoques alternativos para demostrar la equivalencia sustancial en las notificaciones previas a la comercialización». Su objetivo es establecer una vía eficiente para la presentación de solicitudes FDA (k) FDA que incluya ciertos cambios en la solicitud 510(k) ya aprobada. Esta nueva notificación 510(k) ofrece tres tipos de solicitudes: 510(k) especial, 510(k) abreviada y 510(k) tradicional. En 2019,FDA US publicó un documento de orientación especial 510(k) en el que se describe una vía opcional para los fabricantes que realizan determinadas modificaciones bien definidas en sus dispositivos comercializados legalmente.
¿Por qué un 510(k) especial?
Cuando un fabricante desea la aprobación de las modificaciones que ha realizado en el dispositivo ya comercializado, es decir, el dispositivo existente, puede solicitar una 510(k) especial. Los principales factores que hay que tener en cuenta para determinar si una modificación de un dispositivo existente puede ser adecuada para un 510(k) especial son los siguientes:
- El cambio se produce en el propio dispositivo predicado legalmente comercializado del remitente.
- No se requieren datos de rendimiento, o se dispone de métodos bien establecidos si se considera necesario evaluar el cambio.
- Todos los datos de rendimiento en apoyo de una determinación de equivalencia sustancial pueden revisarse en formato de resumen o de análisis de riesgos.
Documentos necesarios para el 510(k) especial
- Carta de presentación
- El nombre del dispositivo legalmente comercializado (existente) del fabricante y el número 510(k)
- Una descripción detallada de los cambios introducidos en el producto que han dado lugar a la presentación de un nuevo formulario 510(k).
- Una comparación del dispositivo modificado con el dispositivo borrado en un formato tabular.
- Otros cambios en el etiquetado o el diseño
- Un resumen conciso de las actividades de control del diseño
- Basándose en el análisis de riesgos, una identificación de las actividades de verificación y/o validación necesarias para cumplir con 21 CFR 820.30
- Formulario de indicaciones de uso
- Una declaración de que el remitente ha cumplido y no incumple actualmente los requisitos del procedimiento de control del diseño, tal como se especifica en 21 CFR 820.30, y que los registros están disponibles para su revisión previa solicitud.
Calendario especial para la revisión 510(k) por parte de laFDA US
Según las directrices de FDA"Refuse to Accept Policy for 510(k)s", el plazo de revisión de las presentaciones especiales 510(k) es de treinta (30) días a partir de su recepción.
¿Cuándo solicitar un 510(k) especial?
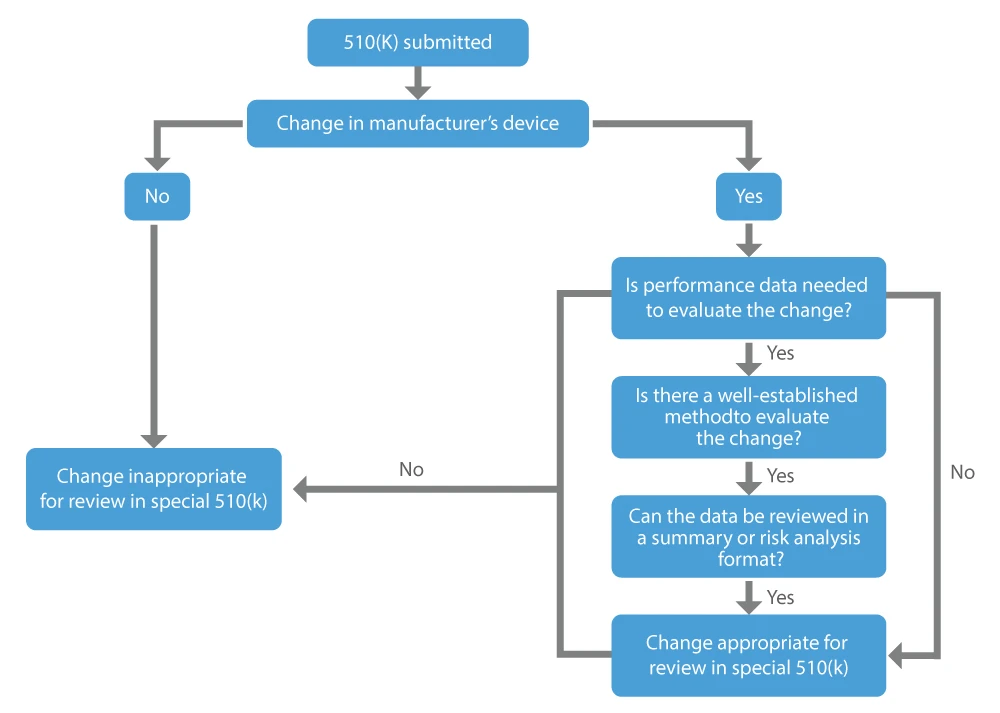
FDA US FDA esfuerzos constantes para proporcionar dispositivos médicos seguros y eficaces que promuevan la salud humana. El programa especial 510(k) es eficiente y coherente con el procedimiento de revisión menos gravoso, que ayuda a los fabricantes extranjeros a vender sus dispositivos en los Estados Unidos y permite a los pacientes acceder oportunamente a nuevos dispositivos médicos.
Para cualquier aclaración adicional sobre el proceso especial 510(k) de FDA, póngase reach contacto con Freyr , un experto acreditado en normativa. Manténgase informado. Cumpla la normativa.