
3 minutes de lecture
Depuis le 31 janvier 2022, la nouvelle réglementation pharmaceutique de l'Union européenne (UE) sur les essais cliniques (CTR) est devenue obligatoire, abrogeant la directive 2001/20/CE sur les essais cliniques. Ce règlement harmonise les protocoles d'évaluation et de supervision des essais cliniques dans l'ensemble de l'UE. Les lignes directrices ont été révisées afin de promouvoir une approche uniforme de la recherche clinique tout en mettant l'accent sur la sécurité des participants aux essais cliniques et sur une meilleure information du public.
Le règlement établit un nouveau système d'évaluation en deux parties pour tous les essais cliniques de l'UE. La première partie consiste en une évaluation scientifique des documents de base de l'essai clinique et la seconde en une évaluation éthique de la documentation au niveau national. À l'issue de cette évaluation en deux parties, chaque État membre prendra une décision unifiée sur l'essai et en informera le promoteur par l'intermédiaire du système d'information sur les essais cliniques.
Délais de transition pour les nouveaux demandeurs
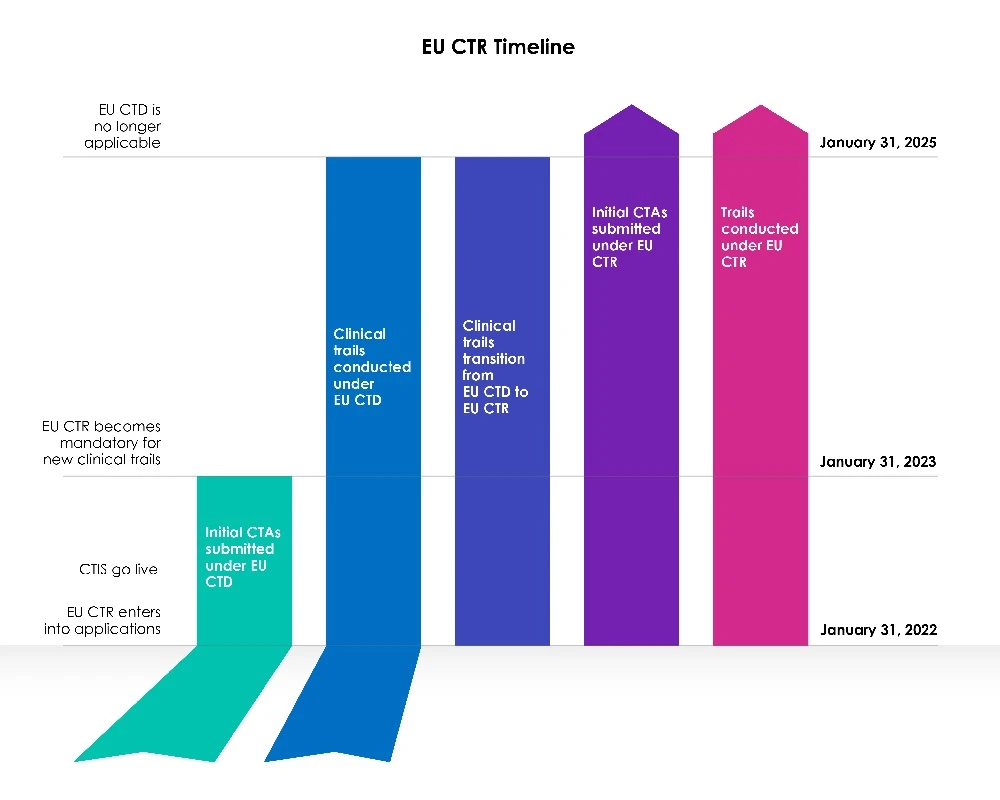
Une phase de transition de trois (03) ans a débuté à la date CTIS de l'UE.
Année 1 (du 31 janvier 2022 au 30 janvier 2023) :
La directive européenne 2001/20/CE relative aux essais cliniques (EU-CTD) régit les essais cliniques dans l'Union européenne depuis 2004. Elle visait à harmoniser les règles et à améliorer considérablement la sécurité des patients dans le cadre des essais cliniques. Cependant, dans la pratique, elle a eu des conséquences imprévues. Au cours de la première année qui a suivi la CTIS , les promoteurs ont eu le choix entre demander une nouvelle autorisation d'essai clinique (CTA) dans le cadre du système d'information sur les essais cliniques (CTIS) prévu par la directive sur les essais cliniques (CTD : directive 2001/20/CE) ou utiliser CTIS à la législation en vigueur, à savoir le règlement (UE) n° 536/2014 sur les essais cliniques.
Les deux idées étaient viables et les sponsors ont eu la possibilité de choisir la législation à poursuivre.
Les membres étaient prêts à utiliser le système d'information sur les essais cliniques (CTIS) et ont accepté les demandes au titre de la nouvelle législation, le règlement sur les essais cliniques (EU CTR), dès le premier jour de CTIS .
Années 2 et 3 (du 31 janvier 2023 au 31 janvier 2025) :
À compter du 31 janvier 2023, toutes les nouvelles demandes de CT devront être soumises via le CTIS la nouvelle législation (CTR).
Les nouvelles demandes CT ne peuvent pas être soumises dans EudraCT en vertu de la directive sur les essais cliniques (CTD). La directive européenne sur les essais cliniques n'autorise plus l'adhésion de nouveaux Member States le 31 janvier 2023. Les essais menés dans le cadre de la CTD doivent d'abord faire l'objet d'une transition, après quoi une demande supplémentaire concernant les États membres peut être soumise via l'EU CTIS.
Pour les demandeurs actuels
Les demandes de tomodensitométrie soumises avant le 30 janvier 2023 en vertu de l'ancienne législation (CTD) utilisant EudraCT, seront autorisées à se poursuivre jusqu'à l'achèvement de cette directive (CTD : directive 2001/20/CE), jusqu'au 30 janvier 2025. Les procédures resteront inchangées et les promoteurs pourront soumettre des modifications significatives et des avis de fin d'essai comme l'exige le règlement. EudraCT restera actif pendant la période de transition afin de permettre la poursuite de ces essais.
Il est toutefois important de noter que les demandes de transition peuvent être déposées à tout moment au cours de la période de transition de trois (03) ans, et les promoteurs sont encouragés à achever le processus suffisamment tôt au cours de la période de transition pour assurer la continuité des essais cliniques de l'UE au-delà du 30 janvier 2025, compte tenu des jours fériés et des deux (02) semaines d'arrêt de l'horloge hivernale.
Essais non transférables
- Les essais qui sont actuellement terminés ou qui s'achèveront juste avant la fin de la périodeEEA ne doivent pas faire l'objet d'une transition.
- Si une notification de fin d'essai a été effectuée dans tous les paysEEA , mais que la fin globale de l'essai n'a pas encore été notifiée, l'étude ne doit pas être transférée. Conformément à la directive, la fin globale de l'essai et le résumé des résultats de l'essai doivent être publiés via EudraCT.
- Les essais qui ont débuté avant la mise en œuvre de la directive 2001/20/CE ne bénéficient pas d'une telle procédure de transition. S'ils sont interventionnels et doivent continuer à fonctionner après la fin de la phase de transition du CTR, une nouvelle demande d'EC doit être émise dans le cadre du CTR.
- Les essais pédiatriques menés en dehors deEEA auxquels un numéro EudraCT a été attribué ne doivent pas non plus être convertis.
- Les essais qui sont suspendus après la fin de la période de transition ne peuvent pas être transférés. La reprise de l'essai dans ces circonstances nécessiterait la soumission d'une nouvelle demande au titre du RTC.
CTIS de l'UE CTIS EMA des mises à jour techniques de EMA afin d'améliorer ses caractéristiques et ses fonctionnalités. Lorsque des modifications importantes sont apportées au CTIS, EMA des notes de mise à jour décrivant les changements apportés au système. Les mises à jour peuvent inclure des améliorations des caractéristiques et fonctionnalités existantes, l'ajout de nouvelles caractéristiques et des améliorations fonctionnelles et techniques. Un partenaire réglementaire expérimenté peut relever les défis potentiels et aider les promoteurs à assurer la transition des essais existants et futurs dans le cadre des stratégies de développement clinique. Cliquez ici pour en savoir plus sur le CTIS l'expertise Freyrdans ce domaine : https://regulatoryaffairs.freyrsolutions.com/clinical-trial-applications-ctas.









