
3 minutes de lecture
La plupart des organisations du secteur des sciences de la vie ont du mal à rationaliser leur processus de soumission réglementaire en raison des changements fréquents apportés aux directives de soumission. Selon une analyse récente menée par la Food and Drug Administration (USFDA) des États-Unis, 32 % des soumissions accompagnées de données d'étude présentaient des problèmes critiques de conformité des données. Le processus nécessite une compréhension approfondie de la collaboration entre les parties prenantes internes et externes. Au sein de l'organisation, les départements de fabrication, de recherche et développement, de recherche clinique/non clinique, QA, de marketing et de vente doivent travailler en harmonie afin d'accélérer le processus d'autorisation, depuis le développement initial jusqu'à la mise sur le marché du médicament. Le département des affaires réglementaires et les autorités sanitaires (HA) sont en contact permanent afin de garantir et d'évaluer la sécurité et l'efficacité des médicaments destinés à l'usage humain. us quelles mesures préliminaires les organisations pharmaceutiques doivent prendre pour mettre en place un processus de soumission réglementaire efficace :
Affecter et déployer des ressources pour rationaliser, normaliser et accélérer le travail de soumission désorganisé.
Un spécialiste de la soumission participe sur une base contractuelle et peut comprendre plus d'un (01) type de soumission tout en étant associé à des programmes de développement au fil des ans, en aidant à rédiger des récits convaincants pour le dossier. Ce faisant, il facilite la tâche des organisations :
- Éliminer les erreurs en équilibrant la collaboration entre les parties prenantes internes et externes
- Réduire le cycle de révision de l'AP du processus de rédaction et d'approbation des documents
- Établir des soumissions complètes et conformes pour suivre l'évolution des exigences réglementaires des différents marchés.
- Accuser réception des soumissions affectées par une autorité de régulation
- Connaître l'état d'avancement de la rédaction, de l'examen et de l'approbation des soumissions
- Amélioration du protocole de soumission actuel
Simplifier la méthodologie de soumission des dossiers
La rédaction d'un dossier est cruciale lorsqu'il s'agit de préparer des soumissions. On observe que les demandes de licence de produits biologiques (BLA) et les demandes de nouveaux médicaments (NDA) comprennent des tonnes de pages, ce qui rend difficile la compréhension de la narration du dossier par les régulateurs. Le fait de disposer d'une représentation structurée d'une idée générale peut renforcer la capacité de l'équipe à préparer les demandes et les agences qui les examinent.
L'utilisation de modules pour rationaliser les soumissions permet aux experts d'analyser ce qui doit être inclus dans chaque section ou module du dossier. Une telle pratique facilite :
- Lancement précoce de la rédaction en accélérant la préparation de la soumission par l'organisation
- Utilisation des données pertinentes nécessaires à l'élaboration des soumissions
- Utilisation des ressources appropriées sans avoir à rédiger des documents exhaustifs à partir de zéro pour chaque soumission.
Travailler en collaboration avec les autorités réglementaires
Chaque organisation doit organiser des réunions régulières avec les AP au cours des premières étapes du développement du produit. Cela permet de garantir un examen et un retour d'information en temps voulu, ce qui accélère la résolution des commentaires par l'AP et, par conséquent, le processus d'approbation. Cela permet non seulement d'établir de bonnes relations entre les organisations et les AP, mais aussi de clarifier les choses à un stade précoce de la planification de la demande d'autorisation.
Rationaliser le contenu et les données dans le système de gestion des documents (SGD)
Un référentiel de contenu robuste doit pouvoir traiter toutes les données de tous les domaines fonctionnels, y compris la documentation et les informations créées précédemment. Un référentiel de contenu et de données structuré permet d'accéder de manière transparente à des documents provenant de divers domaines fonctionnels sans effort supplémentaire ni duplication. Les documents stockés dans le DMS deviennent la source unique de contact pour toutes les parties prenantes internes (science clinique, CMC, non clinique, pharmacocinétique, pharmacodynamique, rédaction médicale et opérations cliniques). Ce contenu et ces données peuvent être utilisés pour créer du contenu local en fonction des exigences régionales. Optimisation des soumissions réglementaires grâce à la gestion structurée du contenu et des données
Adopter la technologie et l'automatisation pour réduire le travail répétitif et les erreurs
Les organisations peuvent utiliser la technologie en recourant à la mise à jour automatique et au contrôle de la qualité des tableaux, listes et figures (TLF) dans le texte par rapport aux données sources afin d'accélérer le processus de documentation. Ainsi, chaque fois qu'une modification est apportée aux tableaux sources existants, une mise à jour est effectuée dans le texte des tableaux, listes et figures. Cela permet de réduire le nombre d'heures de travail, de gagner du temps sur le chemin critique et d'améliorer la précision des données.
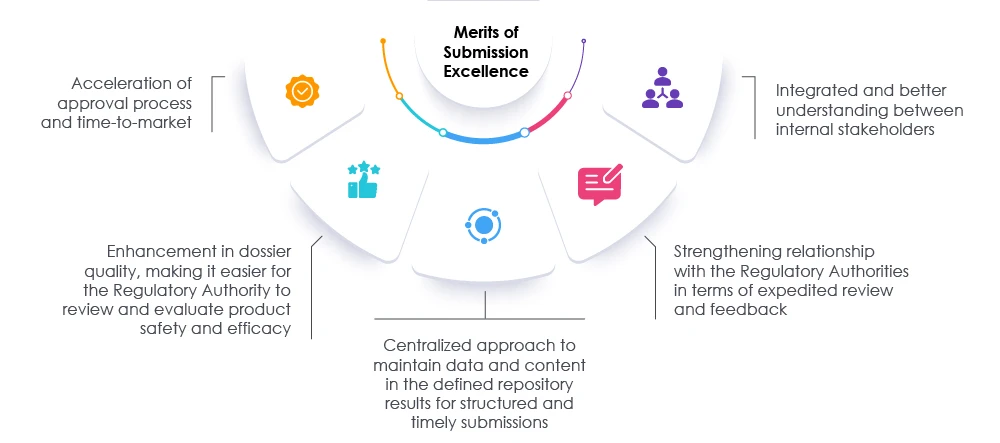
L'adoption de mesures appropriées, associée à un examen rapide des dossiers et des données, peut aider les organismes de réglementation et les organisations à accélérer le processus de soumission. Cette approche accélère la mise sur le marché afin de répondre aux besoins des patients et de l'industrie. Un partenaire réglementaire éprouvé peut aider les organisations à repenser, réévaluer et modifier les approches actuelles en matière de préparation des soumissions. Les experts de Freyr faciliter l'obtention de soumissions réglementaires d'excellence en améliorant la visibilité au sein de l'organisation et en favorisant la conformité globale, permettant ainsi à l'organisation de réussir sur le marché actuel des sciences de la vie. Consultez Freyr.









