
3 min read
Z dniem 31 stycznia 2022 r. nowe rozporządzenie farmaceutyczne Unii Europejskiej (UE) dotyczące badań klinicznych (CTR) stało się obowiązkowe, uchylając dyrektywę w sprawie badań klinicznych 2001/20/WE. Rozporządzenie harmonizuje protokoły oceny i nadzoru badań klinicznych w całej UE. Wytyczne zostały zmienione w celu promowania jednolitego podejścia do badań klinicznych, przy jednoczesnym podkreśleniu bezpieczeństwa uczestników badań klinicznych i zwiększonej jawności publicznej.
Rozporządzenie ustanawia nowy dwuczęściowy system oceny wszystkich badań klinicznych w UE. Część I składa się z naukowej oceny podstawowej dokumentacji badania klinicznego, a część II składa się z etycznej oceny dokumentacji na poziomie krajowym. W następstwie tej dwuczęściowej oceny każde państwo członkowskie podejmie jednolitą decyzję w sprawie badania i powiadomi sponsora za pośrednictwem systemu informacji o badaniach klinicznych.
Terminy przejścia dla nowych wnioskodawców
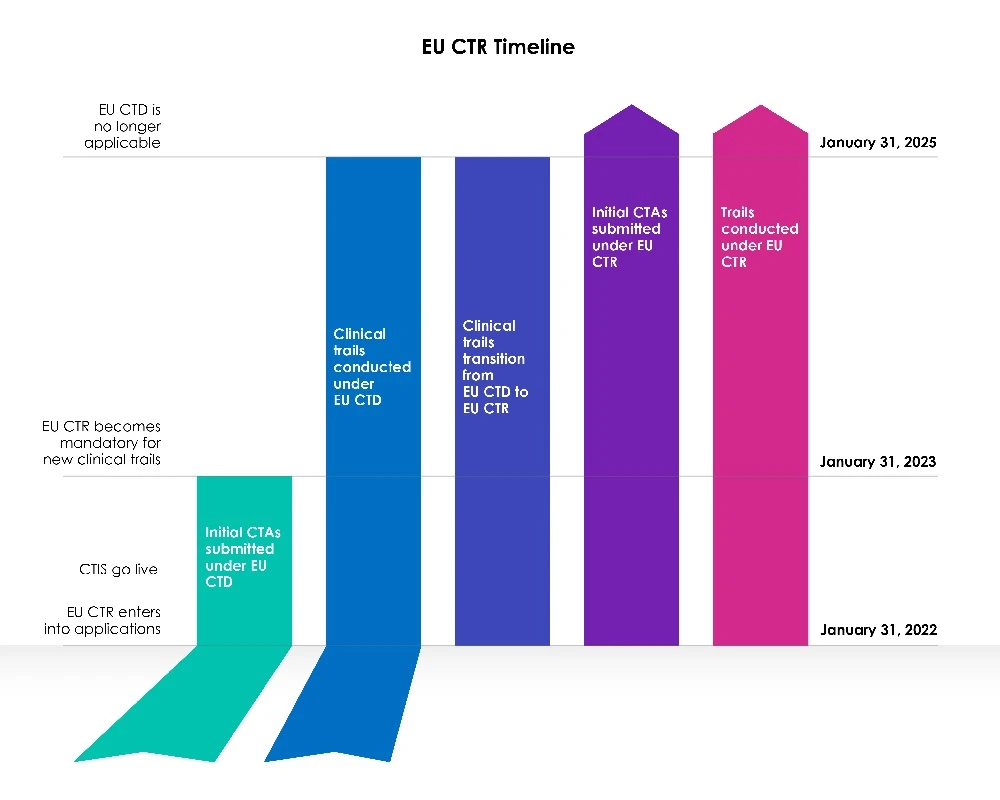
Wraz z datą CTIS systemu CTIS UE rozpoczęła się trzyletnia (03) faza przejściowa.
Rok 1 (od 31 stycznia 2022 r. do 30 stycznia 2023 r.):
Dyrektywa Unii Europejskiej (UE) w sprawie badań klinicznych 2001/20/WE (EU-CTD) reguluje badania kliniczne w UE od 2004 r. Jej celem było ujednolicenie zasad i znaczna poprawa bezpieczeństwa pacjentów uczestniczących w badaniach klinicznych. Jednak w praktyce spowodowała ona niezamierzone konsekwencje. W pierwszym roku po CTIS sponsorzy mogli zdecydować, czy złożyć nowy wniosek o przeprowadzenie badania klinicznego (CTA) w ramach systemu informacji o badaniach klinicznych (CTIS) zgodnie z dyrektywą w sprawie badań klinicznych (CTD: dyrektywa 2001/20/WE), czy też korzystać CTIS z obowiązującymi przepisami, tj. rozporządzeniem w sprawie badań klinicznych (UE) nr 536/2014.
Oba pomysły były wykonalne, a sponsorzy mieli szansę wybrać, które przepisy będą realizowane.
Członkowie byli gotowi do korzystania z systemu informacji o badaniach klinicznych (CTIS) i przyjmowali wnioski zgodnie z nowymi przepisami, rozporządzeniem w sprawie badań klinicznych (EU CTR), już pierwszego dnia CTIS .
Lata 2 i 3 (od 31 stycznia 2023 r. do 31 stycznia 2025 r.):
Od 31 stycznia 2023 r. wszystkie nowe wnioski CT muszą być składane za pośrednictwem systemu CTIS nowymi przepisami (CTR).
Zgodnie z dyrektywą w sprawie badań klinicznych (CTD) nie można składać nowych wniosków CT w systemie EudraCT. Dyrektywa UE w sprawie badań klinicznych nie zezwala już na przyjmowanie nowych Member States 31 stycznia 2023 r. Badania prowadzone zgodnie z dyrektywą CTD muszą najpierw przejść proces transformacji, po czym można złożyć dodatkowy wniosek dotyczący państwa członkowskiego za pośrednictwem CTIS EU CTIS.
Dla obecnych wnioskodawców
Wnioski o CT złożone przed 30 stycznia 2023 r., zgodnie ze starymi przepisami (CTD) z wykorzystaniem EudraCT, będą mogły być rozpatrywane do czasu ich zakończenia zgodnie z tą dyrektywą ((CTD: dyrektywa 2001/20/WE), do 30 stycznia 2025 roku. Procedury pozostaną niezmienione, a sponsorzy będą mogli przedkładać istotne modyfikacje i zawiadomienia o zakończeniu badania zgodnie z wymogami rozporządzenia. EudraCT pozostanie aktywny w okresie przejściowym, aby umożliwić kontynuację tych badań.
Należy jednak zauważyć, że wnioski przejściowe można składać w dowolnym momencie w trakcie trzyletniego (03) okresu przejściowego, a sponsorów zachęca się do zakończenia procesu na tyle wcześnie w okresie przejściowym, aby zapewnić ciągłość badań klinicznych w UE po 30 stycznia 2025 r., biorąc pod uwagę ustawowe święta i dwutygodniowe (02) zimowe zatrzymanie zegara.
Próby niezbywalne
- Badania, które zostały już zakończone lub zostaną zakończone tuż przed końcem okresuEEA , nie mogą zostać przeniesione.
- Jeśli we wszystkich krajachEEA złożono powiadomienie o zakończeniu badania, ale nie zgłoszono jeszcze globalnego zakończenia badania, nie należy przechodzić do kolejnego etapu badania. Zgodnie z dyrektywą globalne zakończenie badania i podsumowanie wyników badania należy opublikować za pośrednictwem systemu EudraCT.
- Badania rozpoczęte przed wdrożeniem dyrektywy 2001/20/WE nie korzystają z takiej procedury przejściowej. Jeśli są to badania interwencyjne i muszą być kontynuowane po zakończeniu fazy przejściowej CTR, należy złożyć nowy wniosek o CT w ramach CTR.
- Badania pediatryczne przeprowadzone pozaEEA którym nadano numer EudraCT, również nie powinny być przekształcane.
- Badania wstrzymane po zakończeniu okresu przejściowego nie mogą zostać przeniesione. Ponowne rozpoczęcie badania w takich okolicznościach wymagałoby złożenia przez CT Application nowego wniosku w ramach CTR.
EU CTIS EMA CTIS aktualizacje techniczne od EMA w celu ulepszenia swoich funkcji i funkcjonalności. W przypadku wprowadzenia istotnych zmian w systemie CTIS, EMA informacje o aktualizacji, w których opisuje zmiany wprowadzone w systemie. Aktualizacje mogą obejmować ulepszenia istniejących funkcji i funkcjonalności, dodanie nowych funkcji oraz ulepszenia funkcjonalne i techniczne. Doświadczony partner w zakresie regulacji prawnych może zająć się potencjalnymi wyzwaniami i pomóc sponsorom w przejściu na nowe badania w ramach strategii rozwoju klinicznego. Kliknij tutaj, aby dowiedzieć się więcej o systemie CTIS doświadczeniu Freyrw tej dziedzinie: https://regulatoryaffairs.freyrsolutions.com/clinical-trial-applications-ctas.









