
3 min de lectura
A partir del 31 de enero de 2022, la nueva normativa farmacéutica de la Unión Europea (UE) para la Regulación de Ensayos Clínicos (CTR) ha pasado a ser obligatoria, derogando la Directiva de Ensayos Clínicos 2001/20/CE. El reglamento armoniza los protocolos de evaluación y supervisión de los ensayos clínicos en toda la UE. Las directrices se revisaron para promover un enfoque uniforme de la investigación clínica, al tiempo que se hace hincapié en la seguridad de los participantes en los ensayos clínicos y en una mayor divulgación pública.
El Reglamento establece un nuevo sistema de evaluación en dos partes para todos los ensayos clínicos de la UE. La Parte I consiste en una evaluación científica de la documentación básica del ensayo clínico, y la Parte II en una evaluación ética de la documentación a nivel nacional. Tras esta evaluación en dos partes, cada Estado miembro tomará una decisión unificada sobre el ensayo y lo notificará al promotor a través del Sistema de Información sobre Ensayos Clínicos.
Plazos de transición para nuevos solicitantes
Con la fecha CTIS de la UE comenzó una fase de transición de tres (03) años.
Año 1 (del 31 de enero de 2022 al 30 de enero de 2023):
La Directiva 2001/20/CE de la Unión Europea (UE) sobre ensayos clínicos (EU-CTD) regula los ensayos clínicos en la UE desde 2004. Su objetivo era estandarizar las normas y mejorar significativamente la seguridad de los pacientes en los ensayos clínicos. Sin embargo, en la práctica, tuvo consecuencias no deseadas. Durante el primer año, tras CTIS , se permitió a los patrocinadores elegir entre solicitar una nueva solicitud de ensayo clínico (CTA) en el marco del Sistema de Información sobre Ensayos Clínicos (CTIS) en virtud de la Directiva sobre ensayos clínicos (CTD: Directiva 2001/20/CE), o utilizar CTIS conformidad con la legislación vigente, el Reglamento (UE) n.º 536/2014 sobre ensayos clínicos.
Ambas ideas eran viables, y los patrocinadores tuvieron la oportunidad de elegir qué legislación seguir.
Los miembros estaban preparados para utilizar el Sistema de Información sobre Ensayos Clínicos (CTIS) y aceptaron solicitudes en virtud de la nueva legislación, el Reglamento sobre ensayos clínicos (EU CTR), el primer día de CTIS .
Años 2 y 3 (del 31 de enero de 2023 al 31 de enero de 2025):
A partir del 31 de enero de 2023, todas las nuevas solicitudes de CT deberán presentarse a través del CTIS la nueva legislación (CTR).
Las nuevas solicitudes de ensayos clínicos no pueden presentarse en EudraCT en virtud de la Directiva sobre ensayos clínicos (CTD). La Directiva sobre ensayos clínicos de la UE ya no permite la admisión de nuevos Member States 31 de enero de 2023. Los ensayos realizados en virtud de la CTD deben someterse primero a una transición, tras lo cual se puede presentar una solicitud adicional relativa a los Estados miembros a través CTIS EU CTIS.
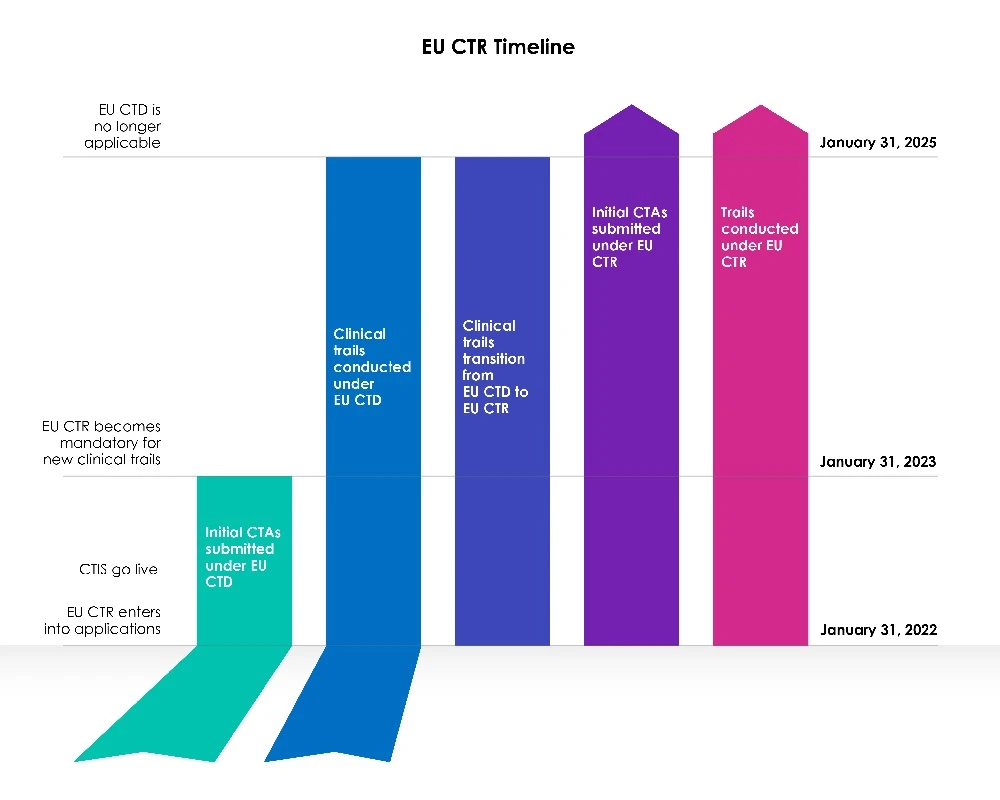
Para los solicitantes actuales
Las solicitudes de TC presentadas antes del 30 de enero de 2023, con arreglo a la antigua legislación (CTD) utilizando EudraCT, podrán ejecutarse hasta la finalización con arreglo a dicha Directiva ((CTD: Directiva 2001/20/CE), hasta el 30 de enero de 2025. Los procedimientos permanecerán inalterados, y los promotores podrán presentar modificaciones significativas y notificaciones de fin de ensayo, tal como exige el reglamento. EudraCT permanecerá activo durante el tiempo de transición para permitir que estos ensayos continúen.
Sin embargo, es importante señalar que las solicitudes de transición pueden presentarse en cualquier momento durante el período de transición de tres (03) años, y se anima a los patrocinadores a completar el proceso lo suficientemente pronto en el período de transición para garantizar la continuidad de los ensayos clínicos de la UE más allá del 30 de enero de 2025, teniendo en cuenta los días festivos legales y la parada del reloj de invierno de dos (02) semanas.
Pruebas intransferibles
- Los ensayos que hayan concluido recientemente o que vayan a concluir justo antes del final del períodoEEA no deben ser objeto de transición.
- Si se ha completado la notificación de finalización del ensayo en todos los paísesEEA , pero aún no se ha notificado la finalización global del ensayo, el estudio no debe pasar a la siguiente fase. Según la Directiva, la finalización global del ensayo y el resumen de los resultados del ensayo deben publicarse a través de EudraCT.
- Los ensayos que comenzaron antes de la aplicación de la Directiva 2001/20/CE no se benefician de dicho procedimiento de transición. Si son intervencionistas y deben seguir funcionando una vez finalizada la fase de transición del RTC, deberá emitirse una nueva solicitud de TC con arreglo al RTC.
- Los ensayos pediátricos realizados fuera de laEEA a los que se les ha asignado un número EudraCT, tampoco deben convertirse.
- Los ensayos que queden en suspenso una vez finalizado el periodo transitorio no podrán ser objeto de transición. La reanudación del ensayo en estas circunstancias requeriría la presentación de una nueva solicitud en virtud del RTC.
CTIS de la UE CTIS actualizaciones técnicas EMA de la EMA para mejorar sus características y funcionalidades. Cuando se realizan cambios importantes en el CTIS, la EMA notas de lanzamiento en las que se describen los cambios introducidos en el sistema. Las actualizaciones pueden incluir mejoras de las características y funcionalidades existentes, la incorporación de nuevas características y mejoras funcionales y técnicas. Un socio regulador con experiencia puede abordar los posibles retos y ayudar a los patrocinadores en la transición de los ensayos existentes y futuros como parte de las estrategias de desarrollo clínico. Haga clic aquí para obtener más información sobre el CTIS la experiencia Freyren este ámbito: https://regulatoryaffairs.freyrsolutions.com/clinical-trial-applications-ctas.









