
3 minutes de lecture
Le protocole de validation est défini comme un plan documenté pour tester un dispositif médical afin de confirmer que le processus de production utilisé pour fabriquer le produit répond aux exigences spécifiques de l'utilisateur, aux exigences techniques et aux exigences réglementaires. Ce plan comprend un examen des variables du processus et des limites opérationnelles, ainsi que l'analyse des résultats des essais dans des conditions d'utilisation réelles.
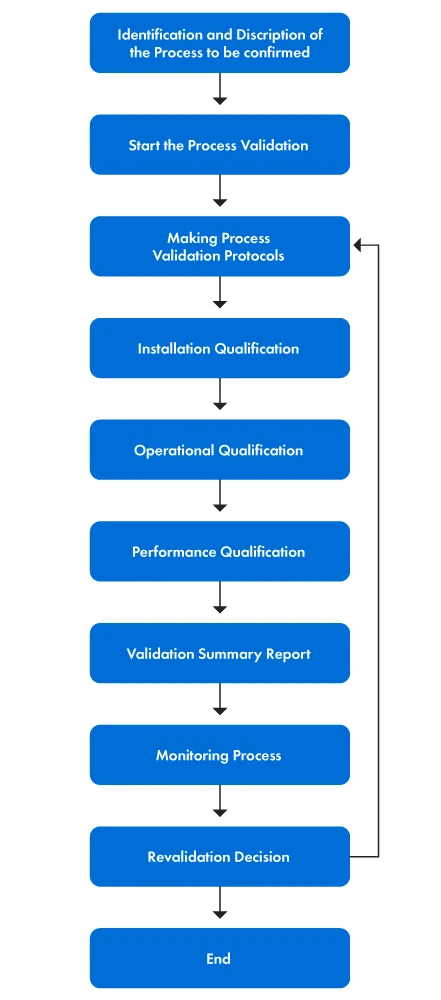
Le processus de validation comporte plusieurs actions concrètes. Les étapes sont expliquées ci-dessous :
- Tout d'abord, l'équipe de validation est constituée et chaque membre se voit attribuer des rôles et des responsabilités spécifiques. Le but de la validation des processus est de fournir une déclaration claire des objectifs de validation et de définir la portée des activités de validation en spécifiant les aspects du dispositif médical qui sont validés. L'équipe comprend ensuite les principes sous-jacents du processus afin d'identifier les paramètres spécifiques et les résultats souhaités.
- Ensuite, les critères d'évaluation et d'acceptation sont établis, de même que la sélection des méthodes d'essai, des outils et des techniques d'analyse statistique appropriés. Ensuite, les protocoles de validation des processus sont rédigés et la qualification de l'installation (QI), la qualification opérationnelle (QO) et la qualification des performances (QP) sont mises en œuvre.
- Enfin, des contrôles continus du processus et des mesures de surveillance sont déterminés pour assurer la validation continue du processus. Si nécessaire, une revalidation est effectuée pour maintenir la précision et l'efficacité du processus de validation.
La figure 1 ci-dessous présente le processus de validation étape par étape.
Figure 1 : Les étapes du processus de validation
PVP
En raison de la grande diversité des volumes de production et de la complexité de la fabrication, il existe de nombreuses approches pour mener à bien la validation des processus. Toutefois, les réglementations de la Food and Drug Administration des États-UnisUSFDA et la ISO 13485 ne fournissent que des suggestions limitées sur des méthodes spécifiques. Néanmoins, un document d'orientation publié en 2004 par le Global Harmonization Task Force (GHTF), aujourd'hui appelé International Medical Device Regulators Forum (IMDRF), constitue une source largement reconnue et faisant autorité en matière de validation des processus de fabrication des dispositifs médicaux. Ce document reste la référence principale, même sur le site officiel de l'USFDA.
Conformément au document d'orientation, une équipe de validation est constituée pour élaborer un plan détaillé de validation des processus (PVP). Les protocoles de validation des procédés comprennent un schéma détaillé sur la manière de mettre en œuvre la QI, la QO, la QP et la revalidation. Le PVP doit contenir les éléments suivants :
- Définir le dispositif et déterminer l'approche de validation.
- Identifier les éléments qui nécessitent une validation.
- Mener des activités sur le site désigné.
- Définir le champ d'application de la documentation.
- Création d'un calendrier pour les activités de validation.
- Élaboration d'un calendrier directeur global.
- Maintenir une liste complète et des références aux validations internes et externes qui ont été effectuées.
Le protocole de validation est rédigé avant de mener les activités de validation. Il doit être préparé par l'équipe de validation et approuvé par le service concerné. L'objectif d'un protocole de validation est de définir les scripts de test à suivre pour garantir que les processus et les équipements sont prêts à fabriquer des dispositifs médicaux sûrs et efficaces.
Un rapport d'analyse contenant des informations ainsi que l'analyse, les explications et les recommandations nécessaires fait partie du protocole de validation. Ces dossiers sont ensuite examinés pour s'assurer que les deux (02) critères suivants sont remplis :
- Respect des normes réglementaires.
- Tous les dossiers et toutes les données générés sont examinés pour en vérifier les résultats, l'adéquation et l'exhaustivité.
La figure 2 ci-dessous représente le PVP et les différents processus qu'il implique.
Figure 2 : Le PVP et ses exigences
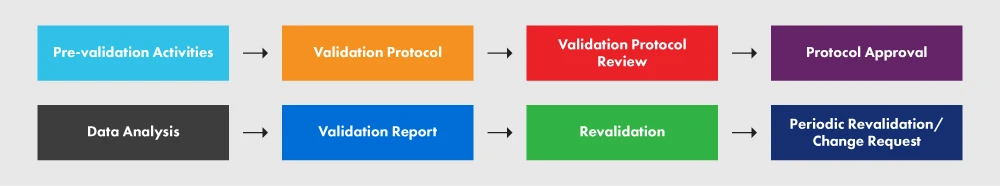
Un protocole correctement rédigé fournit des lignes directrices, des politiques et des procédures claires à respecter pendant la validation du processus. Il englobe des aspects tels que les installations, l'équipement, les méthodes et la formation. Le protocole spécifie les intrants et les limites du processus, ainsi que les étapes essentielles à la bonne exécution du projet de validation du processus. Bien que le schéma suivant n'englobe pas tous les éléments requis dans votre protocole, il vous donne un aperçu du niveau de détail requis. Nous vous recommandons vivement de suivre le document d'orientation pour une meilleure compréhension du processus.
- Page de titre
- Produits à couvrir
- Équipement/processus à valider
- Général
- Objectifs
- Documents de référence
- Plan de validation
- QI
- OQ
- PQ
- Équipement de mesure et d'essai et étalonnage
- Maintenance des équipements
- Revalidation
- Page d'approbation/signature de l'équipe de validation
La gestion des opérations joue un rôle crucial dans le maintien de performances optimales en surveillant les mesures clés, en examinant les méthodes de travail et les procédures et en prenant des mesures rapides en cas de problème. En cas de problème, il peut s'avérer nécessaire de revalider partiellement ou totalement un processus. Selon la section 820.75(c) du règlement sur le système de qualité (QSR) de l USFDA , la revalidation d'un processus doit être envisagée dans ces circonstances : "Lorsque des changements ou des déviations de processus se produisent, le fabricant doit examiner, évaluer et effectuer une revalidation selon le cas. Ces activités doivent être documentées.
Les déclencheurs possibles de la revalidation des procédés sont les modifications des spécifications, des méthodes, des procédures, des logiciels, des conceptions, des composants clés, la mise à l'échelle des lots, les changements d'emplacement, les changements d'équipement, etc. En outre, la mise en œuvre d'actions correctives et préventives (CAPA) peut également servir de déclencheur à la revalidation des procédés. Les principales raisons de la revalidation sont les suivantes :
- Changements apportés au processus.
- Tendance négative de la qualité, détérioration soudaine de la qualité ou augmentation des réclamations des clients.
- Expansion majeure de la capacité de la ligne.
- Changements dans la conception.
- Changements dans l'emballage des produits.
- Transfert d'un processus vers une autre installation.
- Changements dans la procédure de candidature.
Pour en savoir plus sur les protocoles de validation et leur importance dans le domaine de la fabrication de dispositifs médicaux, us Restez informés ! Restez conforme !









