
3 min read
Protokół walidacji definiuje się jako udokumentowany plan testowania wyrobu medycznego w celu potwierdzenia, że proces produkcyjny stosowany do wytwarzania produktu spełnia określone wymagania użytkownika, techniczne i regulacyjne. Obejmuje to przegląd zmiennych procesowych i ograniczeń operacyjnych oraz analizę wyników testów w rzeczywistych warunkach użytkowania.
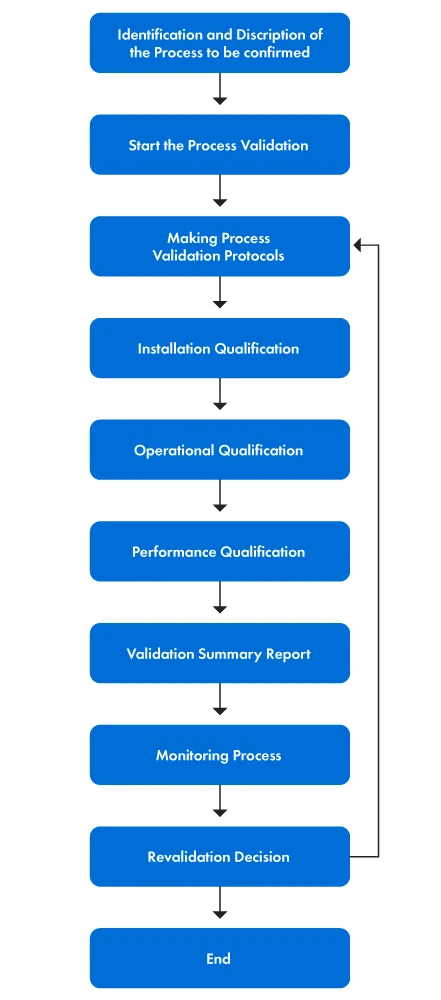
Proces walidacji obejmuje kilka konkretnych działań. Kroki te są wyjaśnione w następujący sposób:
- Najpierw tworzony jest zespół walidacyjny, a każdemu członkowi przypisywane są określone role i obowiązki. Celem walidacji procesu jest jasne określenie celów walidacji i zdefiniowanie zakresu działań walidacyjnych poprzez określenie aspektów urządzenia medycznego, które są walidowane. Następnie zespół rozumie podstawowe zasady procesu, aby zidentyfikować określone parametry i pożądane wyniki.
- Po drugie, ustalane są kryteria oceny i akceptacji, wraz z wyborem odpowiednich metod testowania, narzędzi i technik analizy statystycznej. Następnie opracowywane są protokoły walidacji procesu i wdrażane są kwalifikacje instalacyjne (IQ), kwalifikacje operacyjne (OQ) i kwalifikacje wydajności (PQ).
- Na koniec określane są bieżące kontrole procesu i środki monitorowania w celu zapewnienia ciągłej walidacji procesu. W razie potrzeby przeprowadzana jest ponowna walidacja w celu utrzymania dokładności i skuteczności procesu walidacji.
Poniższy rysunek 1 przedstawia krok po kroku proces walidacji.
Rysunek 1: Etapy procesu walidacji
PVP
Ze względu na szeroki zakres wielkości produkcji i złożoności produkcji, istnieje wiele podejść do przeprowadzania walidacji procesów. Jednak przepisy Amerykańskiej Agencji ds. Żywności i LekówUSFDA) oraz ISO 13485 zawierają ograniczone sugestie dotyczące konkretnych metod. Niemniej jednak, powszechnie uznanym i autorytatywnym źródłem walidacji procesów urządzeń medycznych jest dokument z wytycznymi Global Harmonization Task Force (GHTF), obecnie nazywany International Medical Device Regulators Forum (IMDRF), opublikowany w 2004 roku. Pozostaje on głównym punktem odniesienia nawet na oficjalnej stronie internetowej USFDA.
Zgodnie z wytycznymi, zespół walidacyjny jest tworzony w celu stworzenia szczegółowego Planu Walidacji Procesu (PVP). Protokoły walidacji procesu obejmują szczegółowy schemat wdrażania IQ, OQ, PQ i ponownej walidacji. PVP powinien zawierać następujące elementy:
- Zdefiniowanie urządzenia i określenie podejścia do walidacji.
- Identyfikacja elementów wymagających walidacji.
- Prowadzenie działań w wyznaczonym miejscu.
- Określenie zakresu dokumentacji.
- Tworzenie harmonogramu działań walidacyjnych.
- Opracowanie ogólnego harmonogramu głównego.
- Prowadzenie kompleksowej listy i odniesień do przeprowadzonych walidacji wewnętrznych i zewnętrznych.
Protokół walidacji jest sporządzany przed przeprowadzeniem działań walidacyjnych. Powinien on zostać przygotowany przez zespół walidacyjny i zatwierdzony przez odpowiedni dział. Celem protokołu walidacji jest zdefiniowanie skryptów testowych, których należy przestrzegać, aby zagwarantować, że procesy i sprzęt są gotowe do wytwarzania bezpiecznych i skutecznych wyrobów medycznych.
Raport analityczny, który zawiera informacje wraz z niezbędną analizą, wyjaśnieniami i zaleceniami, jest częścią protokołu walidacji. Zapisy te są dalej weryfikowane w celu zapewnienia, że spełnione są następujące dwa (02) kryteria:
- Spełnienie standardów regulacyjnych.
- Wszystkie zapisy i wygenerowane dane są weryfikowane pod kątem wyników, adekwatności i kompletności.
Poniższy rysunek 2 przedstawia PVP i różne procesy w nim zachodzące.
Rysunek 2: PVP i jego wymagania
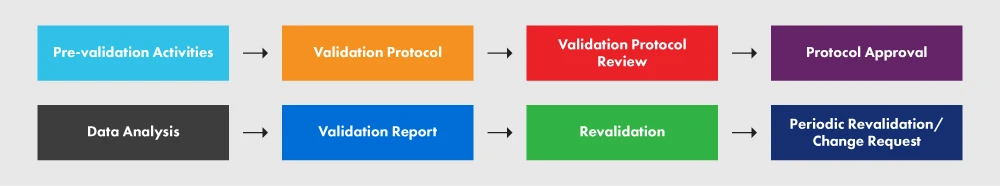
Odpowiednio sporządzony protokół zapewnia jasne wytyczne, zasady i procedury, których należy przestrzegać podczas walidacji procesu. Obejmuje on takie aspekty, jak obiekty, sprzęt, metody i szkolenia. Protokół określa dane wejściowe i limity procesu, a także podstawowe kroki niezbędne do pomyślnej realizacji projektu walidacji procesu. Chociaż poniższy zarys nie obejmuje każdego elementu wymaganego w protokole, daje on przegląd wymaganego poziomu szczegółowości. Zdecydowanie zalecamy przestrzeganie wytycznych w celu lepszego zrozumienia procesu.
- Strona tytułowa
- Produkty objęte ubezpieczeniem
- Sprzęt/proces podlegający walidacji
- Ogólne
- Cele
- Dokumenty referencyjne
- Plan walidacji
- IQ
- OQ
- PQ
- Sprzęt pomiarowy/testujący i kalibracja
- Konserwacja sprzętu
- Rewalidacja
- Strona zatwierdzenia/podpisu zespołu zatwierdzającego
Zarządzanie operacyjne odgrywa kluczową rolę w utrzymaniu optymalnej wydajności poprzez monitorowanie kluczowych środków, przegląd metod pracy i procedur oraz podejmowanie szybkich działań w przypadku pojawienia się jakichkolwiek problemów. W przypadkach, w których występują problemy, może być konieczna częściowa lub nawet pełna ponowna walidacja procesu. Zgodnie z sekcją 820.75(c) rozporządzenia USFDA w sprawie systemu jakości (QSR), w takich okolicznościach należy rozważyć ponowną walidację procesu: "W przypadku wystąpienia zmian lub odchyleń w procesie, producent musi dokonać przeglądu, oceny i odpowiednio przeprowadzić ponowną walidację. Działania te muszą być udokumentowane".
Możliwe czynniki wyzwalające rewalidację procesu obejmują modyfikacje specyfikacji, metod, procedur, oprogramowania, projektów, kluczowych komponentów, skalowanie partii, zmiany lokalizacji, zmiany sprzętu itp. Co więcej, wdrożenie działań naprawczych i zapobiegawczych (CAPA) może również służyć jako czynnik wyzwalający rewalidację procesu. Podstawowe powody rewalidacji są następujące:
- Zmiany wprowadzone do procesu.
- Negatywny trend w zakresie jakości, nagłe pogorszenie jakości lub wzrost liczby skarg klientów.
- Znacząca rozbudowa przepustowości linii.
- Zmiany w projekcie.
- Zmiany w opakowaniach produktów.
- Przeniesienie procesu do innego obiektu.
- Zmiany w procesie aplikacji.
Aby dowiedzieć się więcej o protokołach walidacji i ich znaczeniu w dziedzinie produkcji urządzeń medycznych, skonsultuj się us nami Bądź na bieżąco! Zachowaj zgodność z przepisami!









