3 minutes de lecture
Le règlement européen sur les dispositifs médicaux (EU MDR) 2017/745 définit la portée des dispositifs médicaux stériles comme des dispositifs qui doivent être exempts de micro-organismes (bactéries, virus, etc.). Les dispositifs de classe I ayant une fonction stérile peuvent également obtenir une auto-certification, mais celle-ci diffère en termes d'exigences par rapport aux autres sous-catégories de dispositifs de classe I.
Les exigences relatives aux dispositifs de classe I varient considérablement entre la fabrication et la mise sur le marché. Il en va de même pour les dispositifs médicaux de classe I stériles. Par exemple, si un dispositif de classe I a une fonction stérile, les exigences en matière de fabrication et de mise en place d'un Quality Management System (QMS) très différentes de celles applicables aux dispositifs de classe I ayant des fonctions de mesure.
Cet article vise à fournir une liste de contrôle concise à prendre en compte lors de la mise sur le marché d'un dispositif de classe I en conditions stériles dans l'Union européenne. Cette liste aidera les fabricants à prendre des décisions éclairées concernant différents aspects.
Dos !
Connaître le champ d'application avant la fabrication
Selon le règlement EU MDR, les dispositifs médicaux ayant des fonctions stériles doivent être développés et fabriqués selon des procédures appropriées afin de garantir leur stérilité jusqu'à leur mise sur le marché. Les dispositifs ne peuvent être réutilisés qu'après avoir été soumis à des procédures de nettoyage, de désinfection et de stérilisation appropriées.
Choisir les bonnes pratiques de stérilisation
Il existe différentes méthodes de stérilisation que le fabricant peut choisir pour stériliser le dispositif. La méthode de l'oxyde d'éthylène (EtO) est largement utilisée pour stériliser les dispositifs médicaux. Le choix de la stérilisation doit être justifié de manière appropriée. Les normes EN ISO 14937, ISO TS 21387, ISO TS 13004, ISO 11137 et autres peuvent être suivies par les fabricants pour établir, sécuriser et maintenir la stérilisation.
Il convient également de noter que la documentation relative à la méthode de stérilisation détaillée, à la méthode en cours de fabrication et à la validation des données doit être établie avec précision. Les informations relatives à la stérilisation, telles que la température de chauffage, les conditions ambiantes, les validations de processus, etc., doivent être fournies conformément aux EU MDR .
Connaître les exigences et les orientations à suivre
L'une des étapes cruciales consiste à documenter les exigences techniques et à connaître les normes acceptées dans le cadre EU MDR. Par exemple, la norme EN 556 est la norme européenne relative aux dispositifs stérilisés. La norme EN 556 détaille les exigences relatives à la désignation d'un dispositif médical comme stérile. En outre, la norme ISO TS 19930 spécifie les stratégies permettant de garantir la stérilité des dispositifs. Cependant, elle n'a pas encore été adoptée comme norme européenne.
Connaître son itinéraire
Pour obtenir le marquage CE, il convient de s'informer sur l'intervention des organismes notifiés et sur la procédure d'évaluation de la conformité à suivre. En outre, les dispositifs médicaux stériles doivent respecter les annexes IX et XI.
L'intervention d'un organisme notifié est requise mais limitée. Dans le cas des dispositifs stériles, l'implication des organismes notifiés portera sur le maintien et la garantie des conditions de stérilité. Par exemple, les évaluations de conformité impliqueront des évaluations microbiologiques, des méthodes aseptiques, etc.
À ne pas faire !
Ne manquez pas les exigences en matière d'emballage et d'étiquetage

Il existe des exigences supplémentaires en matière d'emballage et Conformité du labelling les dispositifs stériles. Tout en respectant les exigences en matière d'emballage et d'étiquetage, il ne faut pas oublier les informations qui doivent être apposées. La première étape consiste à apposer un symbole de stérilité (mentionné ci-dessous) sur l'étiquetage du dispositif.
Des informations telles que la déclaration d'état stérile, la méthode de stérilisation, les instructions à suivre si l'emballage stérile est endommagé ou ouvert involontairement, etc. doivent être incluses. La durée de conservation du produit stérile et le processus de restérilisation (le cas échéant) doivent également être indiqués. Le numéro d'identification d'un organisme notifié doit également être apposé sur le dispositif.
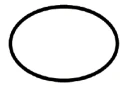
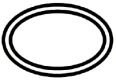
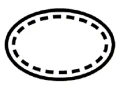
Voici quelques-uns des symboles qui doivent être ajoutés (en fonction de l'applicabilité) dans l'étiquetage des dispositifs médicaux stériles :
Symbole | Indication |
| Dispositifs médicaux stériles dont la méthode de stérilisation est une technique aseptique |
| Dispositifs médicaux stériles dont la méthode de stérilisation est l'oxyde d'éthylène |
| Dispositifs médicaux stériles dont la méthode de stérilisation est la chaleur sèche ou la vapeur. |
| Dispositifs médicaux stériles dont la méthode de stérilisation est le peroxyde d'hydrogène vaporisé |
| Dispositifs médicaux stériles dont la méthode de stérilisation est le rayonnement |
| Le dispositif ne peut pas être stérilisé à nouveau |
| Le dispositif n'est pas stérilisé |
| Ne l'utilisez pas si l'emballage est endommagé |
| Système de barrière stérile unique |
| Double barrière stérile |
| Système de barrière stérile unique avec emballage de protection à l'intérieur |
| Système de barrière stérile unique avec emballage de protection à l'extérieur |


N'oubliez pas les exigences relatives au placement après commercialisation
Les dispositifs stériles doivent tenir un registre des événements indésirables en cas d'occurrence. Dans ce cas, les incidents enregistrés, tels que les documents, doivent être présentés aux autorités compétentes sur demande.
Le EU MDR des exigences spécifiques en matière d'autocertification. Il s'agit d'une procédure complexe, dont le principal facteur réside dans les différentes classes au sein des dispositifs de classe I et leurs différents aspects en matière d'exigences pour l'obtention du marquage CE. Les fabricants de dispositifs de classe I stériles doivent être conscients de la portée et des exigences spécifiques.
Vous souhaitez comprendre en détail les différents aspects des dispositifs stériles de classe I ? Vous aimeriez en discuter avec un expert reconnu ? us.









