
2 min lire
Après la découverte initiale d'impuretés nitrosamines dans des médicaments et des ingrédients pharmaceutiques actifs (APIs) par la USFDA la mi-2018, les organismes de réglementation de l'UE se sont également joints à de nombreux autres pays dans le but de prévenir les risques encourus. Ils ont rappelé plusieurs médicaments présentant des dangers pour la santé liés à cette substance. Connue pour ses propriétés cancérigènes, la nitrosamine peut s'avérer nocive lorsqu'elle est ingérée au-delà des niveaux acceptés par l'homme. La substance, la N-nitrosodiméthylamine (NDMA), a une limite acceptable de 96 ng/jour et tout dépassement de cette limite est inacceptable dans les médicaments et APIs.
Les nitrosamines se forment lorsque des amines secondaires, tertiaires ou quaternaires réagissent avec un agent nitrosant. Tous les médicaments dont les principes actifs sont synthétisés chimiquement font l'objet d'un contrôle visant à détecter la présence d'impuretés. Le Comité des Produits médicaux humain (CHMP) a examiné et rédigé un rapport d'évaluation. Il a demandé aux titulaires d'autorisations de mise sur le marché (AMM) de suivre les dernières recommandations pour examiner tous les médicaments chimiques et biologiques actuellement disponibles sur le marché à destination de la consommation humaine.
Les titulaires d'autorisations de mise sur le marché (MAH) sont chargés de veiller à ce que le processus de fabrication de tous les produits biologiques et chimiques soit régulièrement contrôlé afin de détecter toute contamination. Une fois celle-ci identifiée, les mesures appropriées doivent être prises pour atténuer les risques qu'elle présente. Bien que les risques de contamination soient moindres lors de la fabrication de médicaments chimiques et biologiques, l'Agence européenne des médicaments (EMA) ne prend aucun risque à cet égard. Les entreprises pharmaceutiques doivent s'assurer que les protocoles de fabrication pertinents sont en place, conformément aux dernières EMA . Elles sont également chargées de vérifier les niveaux de nitrosamine dans les médicaments disponibles sur le marché et de les maintenir dans les limites acceptables.
Après avoir finalisé l'année dernière un examen au titre de l'article 5, paragraphe 3, du règlement (CE) n° 726/2004 (demande de réexamen), EMA publié de nouvelles lignes directrices visant à éviter la présence de contamination par les nitrosamines dans les médicaments à usage humain. Le processus est le même pour les produits autorisés au niveau national et ceux autorisés au niveau central. EMA, en collaboration avec la Direction européenne de la qualité du médicament et des soins de santé (EDQM), mettra en œuvre l'article 5, paragraphe 3, du CHMP. Voici la procédure à suivre pour rester en conformité avec les modifications.
L'orientation en trois étapes pour les TAMM
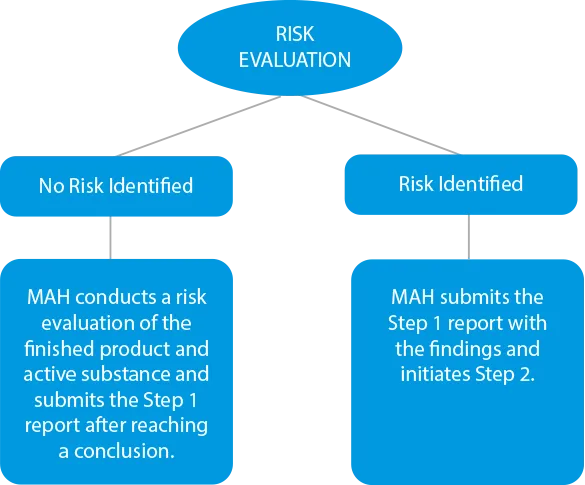
- Évaluation des risques - Les fabricants doivent mener un processus d'évaluation des risques pour identifier les substances actives et le produit fini afin de vérifier les niveaux de nitrosamine. S'il y a des cas de contamination croisée, ils doivent également être inclus dans le rapport de résultats. Les dates limites de soumission à ce stade ont été fixées au 31 mars 2021 pour les médicaments chimiques et au 1er juillet 2021 pour les médicaments biologiques.
![]()
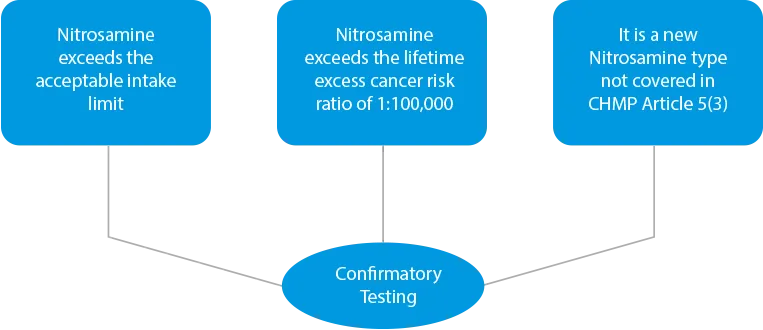
- Tests de confirmation - En cas de contamination croisée ou si des produits sont identifiés comme présentant un risque en raison de la présence de niveaux élevés de nitrosamine, des tests de confirmation doivent être effectués. Les tests de confirmation sont obligatoires dans trois (03) cas.
![]()
- Modifications de l'autorisation de mise sur le marché – Lorsque la présence de nitrosamine est détectée, deux (02) tests de confirmation doivent être effectués afin de communiquer les résultats corrects à EMA. Sur cette base, les titulaires d'autorisation de mise sur le marché doivent demander une modification du processus de fabrication. Pour ce faire, ils doivent suivre les procédures réglementaires standard et modifier le modèle d'autorisation de mise sur le marché. Les délais pour ces procédures sont fixés au 26 septembre 2022 pour les produits chimiques et au 1er juillet 2023 pour les médicaments biologiques.
La voie idéale pour les détenteurs d'AMM pour faire face à la nouvelle conformité de la teneur en nitrosamines
Comme il s'agit de la dernière mesure EMA pour atténuer les risques liés aux niveaux de nitrosamine dans les médicaments et à la contamination croisée, l'ensemble du processus peut s'avérer assez fastidieux pour les titulaires d'autorisations de mise sur le marché et les fabricants d'ingrédients actifs pharmaceutiques. Qu'il s'agisse de soumettre le modèle de réponse lorsque la contamination est détectée à l'étape 1 ou d'effectuer les tests nécessaires, chaque phase doit être conforme aux nouvelles règles. Les titulaires d'autorisations de mise sur le marché doivent collaborer avec des experts en réglementation qui sont au fait de toutes les dernières modifications et veiller à la conformité avec les nouvelles directives. Choisissez le bon partenaire, comme Freyr éviter tout retard et toute erreur.









