
2 min lire
Les dispositifs médicaux font l'objet d'une évaluation de la conformité avant d'être inclus dans la liste ARTG afin de garantir qu'ils sont conformes aux principes essentiels exigés par la Therapeutic Goods Administration (TGA), Australie. Les principes essentiels décrivent les caractéristiques de sécurité et de performance auxquelles tout dispositif doit répondre pour pouvoir être vendu en Australie. Comme dans l'UE, l'évaluation de la conformité en Australie est basée sur la classe de risque du dispositif. Par conséquent, la voie d'évaluation de la conformité à suivre pour tout dispositif dépend de sa classification.
Il existe plusieurs normes réglementaires qui s'appliquent à différents types d'appareils. Le respect d'une ou de plusieurs de ces normes est considéré comme une exigence essentielle. L'identification des normes pertinentes applicables au dispositif et les essais ultérieurs du dispositif pour démontrer sa conformité à la norme deviennent une condition préalable.
L'évaluation de la conformité implique un examen systématique des documents techniques relatifs au dispositif. La gestion des risques, l'évaluation clinique, le processus de fabrication et les activités de vigilance menées par le fabricant sont les domaines critiques à évaluer. Les dispositifs qui doivent obtenir un certificat d'évaluation de la conformité de la TGA sont énumérés dans la réglementation 4.1 du règlement de 2002 sur les produits thérapeutiques (dispositifs médicaux). Si un certificat d'évaluation de la conformité délivré par la TGA est une condition préalable à la commercialisation de la plupart des dispositifs médicaux en Australie, la TGA accepte également les certificats d'évaluation de la conformité délivrés par les organismes notifiés en Europe. En outre, la TGA accepte également les évaluations de conformité des pays qui font partie du MDSAP Medical Device Single Audit Program).
Les exigences en matière d'évaluation de la conformité varient en fonction de la catégorie de risque du dispositif. Le tableau n° 1 détaille le processus d'évaluation de la conformité basé sur la classification en Australie.
Procédure d'évaluation de la conformité - Australie
Classe | Procédure d'évaluation de la conformité | Responsabilités du fabricant |
| I | Partie 6 (Déclaration de conformité ne nécessitant pas d'évaluation par le secrétaire) | Documents attestant de la conformité aux principes essentiels |
| I (mesure) & IIa (non stérile) | Partie 6 (Déclaration de conformité ne nécessitant pas d'évaluation par le secrétaire) Partie 5 (Système de gestion de la qualité des produits) | Documentation démontrant la conformité avec les principes essentiels. Mettre en œuvre le système de gestion de la qualité des produits pour l'inspection finale et les essais en vue des audits. Note : La présentation d'une déclaration de conformité n'est pas requise pour la classe I (non mesurable et non stérile), mais doit être disponible sur demande de la TGA. |
| I (stérile) & IIa (stérile) | Partie 6 (Déclaration de conformité ne nécessitant pas d'évaluation par le secrétaire) Partie 4 (Assurance de la qualité de la production) | Documentation démontrant la conformité avec les principes essentiels. Mettre en œuvre un système de gestion de la qualité qui exclut les éléments de conception, basé sur ISO 13485. |
| IIb | Partie 1 (Assurance qualité complète) à l'exclusion de la clause 1.6 (Examen de la conception) | Mettre en œuvre un système complet de gestion de la qualité, incluant la conception, la production, l'étiquetage, l'emballage et l'inspection finale, conformément à ISO 13485. À l'exclusion du dossier de conception. |
| III & AIMD | Partie 1 (Assurance qualité complète), y compris la clause 1.6 (Examen de la conception) | Mettre en œuvre un système complet de gestion de la qualité, incluant la conception, la production, l'étiquetage, l'emballage et l'inspection finale, conformément à ISO 13485. Concevoir le dossier conformément aux principes essentiels. |
| Paquets de systèmes ou de procédures Partie 7 | (Procédures relatives aux dispositifs médicaux utilisés à des fins particulières) | Principes essentiels. Procédure d'évaluation de la conformité. Preuves cliniques pour chaque composant d'un système ou d'un emballage. |
Tableau n° 1. Exigences en matière d'évaluation de la conformité des dispositifs
La procédure habituelle d'approbation des dispositifs médicaux par la TGA se déroule comme suit :
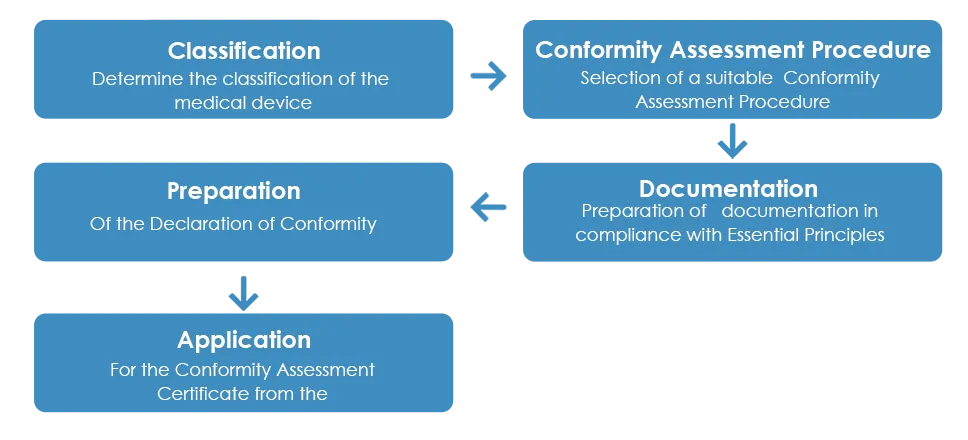
Comme indiqué ci-dessus, une fois le certificat d'évaluation de la conformité reçu de la TGA, une déclaration de conformité (DoC) doit être préparée par le fabricant, déclarant que le dispositif médical est conforme aux principes essentiels applicables, aux règles de classification et à la procédure d'évaluation de la conformité. Toutefois, contrairement au certificat d'évaluation de la conformité, une DoC européenne n'est pas acceptée par la TGA.
Le marché australien offre un paysage prometteur aux fabricants de dispositifs médicaux, si les exigences réglementaires de la Therapeutic Goods Administration (TGA) sont respectées. Les exigences de conformité et le processus d'évaluation varient en fonction de la classe de risque du dispositif et du DIV. Les exigences de conformité et le processus d'évaluation varient en fonction de la classe de risque du dispositif et de la DIV. Bien que les réglementations relatives aux dispositifs médicaux soient bien définies et transparentes, il est complexe de s'y retrouver et les fabricants peuvent demander l'aide des partenaires réglementaires pour obtenir l'inscription de leur dispositif sur la liste.
Pour en savoir plus sur les exigences en matière d'évaluation de la conformité, le parrainage australien et l'inscription à l'ARTG en Australie, contactez un expert en réglementation. Restez informé. Restez en conformité.









