
2 min read
Po pierwszym wykryciu zanieczyszczeń nitrozoaminą w lekach i aktywnych składnikach farmaceutycznych (APIs) przez USFDA połowie 2018 r., organy regulacyjne UE dołączyły do wielu innych krajów w celu zapobiegania związanym z tym zagrożeniom. Wycofały one z rynku kilka leków, które stanowiły zagrożenie dla zdrowia ze względu na obecność tej substancji. Nitrozoamina, znana ze swoich właściwości rakotwórczych, może okazać się szkodliwa dla zdrowia, jeśli zostanie spożyta w ilościach przekraczających dopuszczalne poziomy dla ludzi. Substancja N-nitrozodimetyloamina (NDMA) ma dopuszczalny limit 96 ng/dzień, a wszystko powyżej tej wartości jest niedopuszczalne w lekach i APIs.
Nitrozoaminy powstają w wyniku reakcji amin drugorzędowych, trzeciorzędowych lub czwartorzędowych z czynnikiem nitrozującym. Wszystkie leki zawierające syntetyczne składniki aktywne są sprawdzane pod kątem obecności zanieczyszczeń. Komitet ds. Produkty lecznicze Ludzi (CHMP) dokonał przeglądu i sporządził raport oceniający. Zwrócił się do Pozwolenie na dopuszczenie do obrotu produktu leczniczego (MAH) o przestrzeganie najnowszych wytycznych dotyczących przeglądu wszystkich leków chemicznych i biologicznych dostępnych obecnie na rynku do stosowania u ludzi.
Posiadacze pozwolenia na dopuszczenie do obrotu (MAH) są odpowiedzialni za zapewnienie, że proces produkcji wszystkich produktów biologicznych i chemicznych jest okresowo weryfikowany w celu wykrycia ewentualnego zanieczyszczenia. Po wykryciu zanieczyszczenia należy podjąć odpowiednie kroki w celu wyeliminowania ryzyka, jakie stwarza. Chociaż prawdopodobieństwo wystąpienia takiego zanieczyszczenia jest mniejsze podczas produkcji leków chemicznych i biologicznych, Europejska Agencja Leków (EMA) nie zamierza ryzykować. Firmy farmaceutyczne muszą zapewnić, że stosowane są odpowiednie protokoły produkcyjne zgodnie z najnowszymi EMA . Są one również odpowiedzialne za sprawdzanie poziomu nitrozoamin w lekach dostępnych na rynku i utrzymywanie go w dopuszczalnych granicach.
Po zakończeniu przeglądu zgodnie z art. 5 ust. 3 rozporządzenia (WE) nr 726/2004 (wezwanie do przeglądu) w zeszłym roku, EMA nowe wytyczne dotyczące zapobiegania obecności zanieczyszczeń nitrozoaminami w lekach stosowanych u ludzi. Proces ten jest taki sam dla produktów dopuszczonych do obrotu na poziomie krajowym, jak i centralnym. EMA wraz z Europejską Dyrekcją ds. Jakości Leków i Opieki Zdrowotnej (EDQM) będzie wdrażać art. 5 ust. 3 CHMP. Oto proces, który należy stosować, aby zachować zgodność z modyfikacjami.
Trzyetapowe wytyczne dla MAH
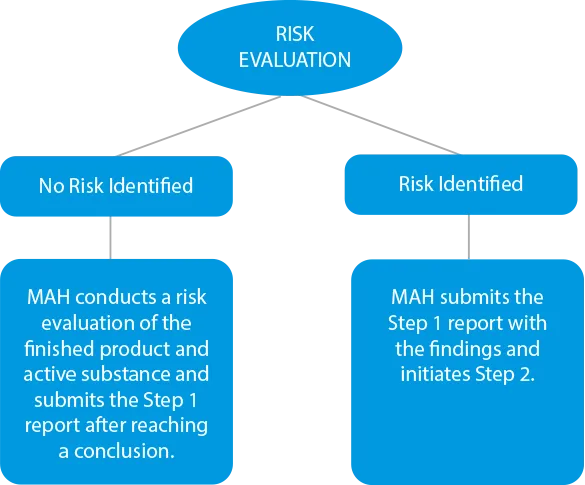
- Ocena ryzyka - Producenci muszą przeprowadzić proces oceny ryzyka, aby zidentyfikować substancje czynne i gotowy produkt w celu sprawdzenia poziomów nitrozoaminy. Jeśli wystąpią jakiekolwiek przypadki zanieczyszczenia krzyżowego, należy je również uwzględnić w raporcie wynikowym. Terminy składania wniosków na tym etapie zostały ustalone na 31 marca 2021 r. dla leków chemicznych i 01 lipca 2021 r. dla leków biologicznych.
![]()
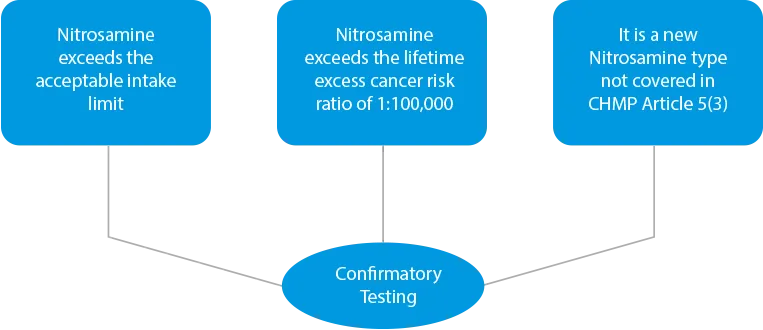
- Testy potwierdzające - W przypadku stwierdzenia jakiegokolwiek zanieczyszczenia krzyżowego lub jeśli produkty zostaną zidentyfikowane jako ryzykowne ze względu na obecność wyższych poziomów nitrozoaminy, należy przeprowadzić testy potwierdzające. Testy potwierdzające są obowiązkowe w trzech (03) przypadkach.
![]()
- Pozwolenie na dopuszczenie do obrotu produktu leczniczego – W przypadku wykrycia obecności nitrozoaminy należy przeprowadzić dwa (02) testy potwierdzające, aby zgłosić prawidłowe odczyty do EMA. Na tej podstawie posiadacze pozwolenia na dopuszczenie do obrotu muszą złożyć wniosek o zmianę procesu produkcyjnego. Odbywa się to przy użyciu standardowych procedur regulacyjnych z wykorzystaniem zmiany do Pozwolenie na dopuszczenie do obrotu produktu leczniczego . Terminy dla tych działań to 26 września 2022 r. dla produktów chemicznych i 1 lipca 2023 r. dla leków biologicznych.
Idealne rozwiązanie dla MAH w zakresie zgodności z nowym poziomem nitrozoaminy
Ponieważ jest to najnowsza propozycja EMA ograniczenie ryzyka związanego z poziomem nitrozoamin w lekach i sytuacją zanieczyszczenia krzyżowego, cały proces może być dość przytłaczający dla posiadaczy pozwolenia na dopuszczenie do obrotu (MAH) i producentów API. Niezależnie od tego, czy chodzi o złożenie szablonu odpowiedzi w przypadku wykrycia zanieczyszczenia na etapie 1, czy o przeprowadzenie kolejnych badań, każdy etap musi być zgodny z nowymi przepisami. Posiadacze pozwolenia na dopuszczenie do obrotu muszą współpracować z ekspertami ds. regulacyjnych, którzy są na bieżąco ze wszystkimi najnowszymi zmianami i zapewniają zgodność z nowymi wytycznymi. Wybierz odpowiedniego partnera, takiego jak Freyr uniknąć opóźnień i błędów.









