2 min lire
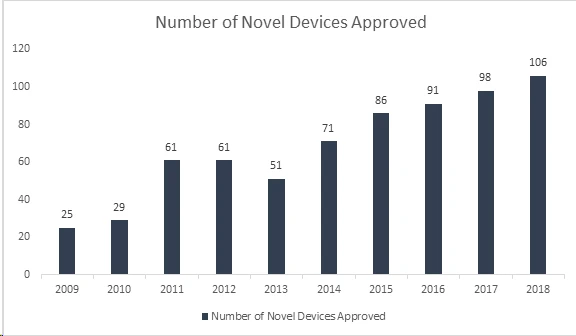
Le saviez-vous ? La US and Drug Administration (FDA) US a établi un nouveau record en approuvant 106 nouveaux dispositifs médicaux en 2018, ce qui en fait l'année la plus fructueuse pour les dispositifs médicaux. Grâce à cette réussite, FDA dépassé son record vieux de 40 ans, établi en 2017 avec l'approbation de 99 nouveaux dispositifs, affichant ainsi une croissance continue depuis 8 ans. Les dispositifs approuvés comprennent une gamme de produits innovants tels que des systèmes automatisés de dosage d'insuline pour les enfants, la plus petite valve cardiaque au monde pour les nouveau-nés, la première application médicale mobile pour aider à gérer les troubles liés à l'abus d'opioïdes et de substances, et des technologies d'intelligence artificielle qui diagnostiquent la rétinopathie diabétique.
FDA toujours encouragé la sécurité et l'innovation des dispositifs médicaux afin de garantir leur haute qualité. Afin de suivre le rythme croissant des autorisations de nouveaux dispositifs et d'assurer leur sécurité, FDA de « moderniser » la procédure d'autorisation des dispositifs médicaux. Selon l'agence, la modernisation de la procédure d'autorisation pourrait nécessiter une nouvelle autorité. En 2018, FDA le Center for Devices and Radiological Health (CDRH) ont publié un document conjoint indiquant que le 510(k) était l'un des deux types de soumission ajoutés à la définition des dispositifs nouveaux. Parallèlement, les exemptions pour dispositifs humanitaires (HDE) ont également été ajoutées à la définition des dispositifs innovants à la suite des modifications apportées au programme du CDRH sur les dispositifs médicaux révolutionnaires en vertu de la loi 21st Century Act.
La proposition de la FDA du CDRH répond au besoin potentiel d'obtenir de nouveaux pouvoirs pour moderniser le processus 510(k). L'objectif de cette proposition est de limiter l'utilisation des dispositifs de référence, considérés comme substantiellement équivalents (SE), qui ont plus de 10 ans, afin de promouvoir l'innovation. Il s'agit d'une avancée par rapport aux directives publiées par l'agence en avril 2018. Le projet a été publié par la FDA proposer l'extension du programme 510(k) abrégé au CDRH de FDAsous le titre « Safety and Performance-based Pathway » (Voie fondée sur la sécurité et la performance). Il a été introduit afin de réduire la charge que représentent les dispositions relatives aux dispositifs médicaux. Cette approche vise également à accroître l'efficacité de l'examen des demandes 510(k), réduisant ainsi la pression exercée sur l'agence.
Voici quelques-uns des points saillants de ces orientations :
- La nouvelle voie d'accès évalue la sécurité et l'efficacité des dispositifs par rapport à des normes de sécurité et à des paramètres de performance définis.
- Malgré les nouvelles normes, les appareils devront se conformer aux normes existantes pour être commercialisés.
- Les technologies modernes seront testées par rapport à des normes modernes
- L'approche favorisera une plus grande concurrence pour la mise au point de dispositifs plus sûrs.
Le nombre de dispositifs médicaux soumis à autorisation a augmenté de manière exponentielle au fil des ans. Cela a donné à la FDA une grande latitude FDA adopter et mettre en œuvre des mesures innovantes visant à améliorer les procédures d'autorisation. L'agence est fermement convaincue du bien-fondé de cette proposition, mais la réaction du secteur reste à déterminer.
La FDA publiant FDA de nouveaux documents d'orientation visant à améliorer l'enregistrement des dispositifs médicaux, les fabricants de dispositifs médicaux doivent se tenir informés et agir en conséquence. Restez à jour. Restez en conformité.









