
5 minutes de lecture
Les logiciels pour appareils médicaux en Corée du Sud sont utilisés pour diagnostiquer, traiter et surveiller les patients dans le système de santé moderne. Ils comprennent à la fois les logiciels intégrés dans les dispositifs médicaux et les logiciels autonomes qui peuvent être utilisés sur les PC, les appareils mobiles et les services basés sur le web. Le ministère sud-coréen de l'alimentation et de la sécurité des médicaments (MFDS) est chargé de réglementer les logiciels de dispositifs médicaux et de garantir leur sécurité et leur efficacité. Le 5 juillet 2023, le MFDS a défini des critères pour l'approbation et l'inspection des logiciels de dispositifs médicaux ; ces règlements fournissent une structure que les requérants civils peuvent suivre lorsqu'ils soumettent un logiciel pour approbation ou examen.
La réglementation aborde divers sujets, notamment le champ d'application, les exigences en matière de documentation technique et les rapports de vérification de la conformité. Outre les directives du MFDS, il existe des normes et des lignes directrices internationales qui s'appliquent aux logiciels pour dispositifs médicaux, telles que la norme 62304 de la Commission électrotechnique internationale (CEI) relative aux processus du cycle de vie des logiciels et les directives de la Food and Drug Administration (FDAUS sur les applications médicales mobiles.
Plan de développement du logiciel et analyse des besoins
- Le plan de développement logiciel décrit l'approche globale du développement logiciel, y compris les spécifications, les méthodes et les outils de développement. Il couvre également la vérification, la gestion des risques liés aux dispositifs médicaux, la gestion de la configuration et la documentation.
- L'analyse des exigences établit les exigences des logiciels de dispositifs médicaux, y compris les mesures de contrôle des risques et les méthodes de vérification. En planifiant et en analysant soigneusement le processus de développement du logiciel, les développeurs peuvent s'assurer que le logiciel qui en résulte répond aux normes de sécurité et d'efficacité nécessaires.
- Le rapport de vérification de la conformité du logiciel comprend un aperçu du plan de développement du logiciel, le numéro de contrôle du document du fabricant et une vue d'ensemble de l'analyse des exigences. En respectant ces lignes directrices, les logiciels de dispositifs médicaux peuvent être développés en toute confiance, sachant qu'ils ont été soumis à des tests rigoureux et qu'ils répondent aux normes de sécurité et d'efficacité nécessaires.
Vérification et validation des logiciels de dispositifs médicaux
- La vérification des logiciels de dispositifs médicaux permet de s'assurer que le logiciel répond aux exigences spécifiées.
- La validation des logiciels de dispositifs médicaux garantit que le logiciel répond aux besoins de l'utilisateur et à l'usage prévu.
- Le rapport de vérification et de validation d'un logiciel de dispositif médical décrit le processus de vérification et de validation, y compris le nom du produit, la révision et les noms des personnes who examiné et approuvé le rapport. Le rapport peut varier en fonction des caractéristiques du logiciel, mais il doit inclure une description du logiciel, les méthodes de vérification et de validation utilisées et les résultats des essais.
Environnement d'exploitation et logiciels de provenance inconnue (SOUP)
- Si le logiciel dépend d'un matériel spécifique, tel qu'un logiciel embarqué, le document technique doit décrire les spécifications du matériel.
- Toutefois, si le logiciel est autonome et développé pour fonctionner sur du matériel polyvalent, l'environnement d'exploitation doit être décrit dans la documentation brute. Cela inclut les spécifications minimales recommandées, telles que Microsoft Windows 10 ou une version supérieure.
- En outre, si le logiciel du dispositif médical comprend un logiciel commercial de provenance inconnue (SOUP), un environnement d'exploitation doit être créé pour garantir son bon fonctionnement. En décrivant soigneusement l'environnement d'exploitation et en tenant compte de tout SOUP, les développeurs peuvent garantir que leur logiciel de dispositif médical est sûr et efficace pour l'usage auquel il est destiné.
Gestion des risques liés aux dispositifs médicaux et exigences en matière de documentation
- Le processus de gestion des risques liés aux software as a medical device comprend l'identification des situations dangereuses, l'établissement de mesures de contrôle des risques, la vérification de ces mesures et la gestion des modifications apportées aux logiciels.
- Le document MFDS-RM sur la gestion des risques logiciels fournit des informations sur la gestion des risques logiciels.
- En outre, les exigences en matière de documentation sont essentielles pour garantir que le logiciel répond aux normes de sécurité et d'efficacité nécessaires.
- Le plan de développement du logiciel, l'analyse des exigences logicielles des dispositifs médicaux et les rapports de vérification et de validation du logiciel doivent être inclus dans la documentation.
- Le rapport de vérification de la conformité du logiciel décrit les exigences en matière de documentation ; il comprend également un aperçu des documents applicables et le numéro de contrôle des documents du fabricant.
Figure 1 : Processus de gestion des risques liés aux dispositifs médicaux
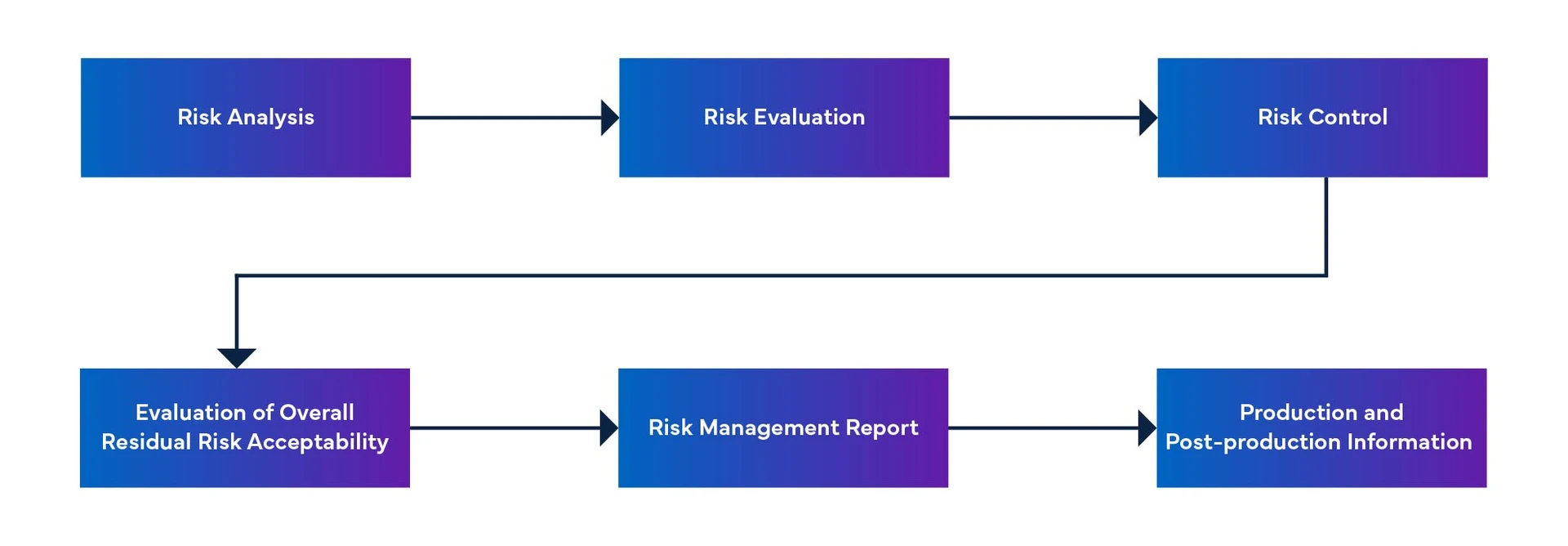
Anomalies non résolues et actions correctives pour le logiciel SaMD
- Le document MFDS-PR (Software Problem Resolution) décrit le processus de résolution des problèmes logiciels, qui comprend le signalement, l'analyse, la mise en œuvre et la vérification des problèmes.
- Le document comprend également une liste des problèmes non résolus, des bogues, des défauts et des anomalies, ainsi qu'une évaluation du risque résiduel pour le système logiciel.
- Les mesures correctives prises pour résoudre ces problèmes doivent être documentées dans le plan de maintenance du logiciel, qui est établi conformément au processus de maintenance du logiciel.
- Le document MFDS-maintenance fournit des informations sur SaMD et le dépannage.
Exigences en matière d'examen et de soumission de documents techniques pour les logiciels SaMD
Les principaux documents examinés au cours de la procédure d'examen sont les données relatives aux performances, le rapport de confirmation de la conformité et les données relatives à la vérification et à la validation du logiciel du dispositif médical, la spécification de conception du logiciel (SDS), la déclaration des exigences du logiciel du dispositif médical (SRS) et les rapports de vérification et de validation. Le rapport de confirmation de la conformité et le rapport de vérification et de validation du logiciel de dispositif médical doivent être soumis.
Gestion des risques liés aux logiciels de dispositifs médicaux
- Identifier les risques potentiels associés au logiciel et à son utilisation.
- Évaluer la gravité des risques associés à ces dangers.
- Mettre en œuvre des mesures de contrôle des risques afin de minimiser la probabilité d'un préjudice.
- Contrôler et évaluer l'efficacité de ces mesures de contrôle des risques.
- Documenter toutes les activités et décisions en matière de gestion des risques liés aux dispositifs médicaux.
Dans un système logiciel, les éléments logiciels sont décomposés en parties plus petites, y compris les éléments logiciels détaillés. Lorsqu'un élément ne peut être décomposé davantage, il est appelé unité. Le système permet une décomposition au niveau de l'unité, ce qui aide à déterminer le niveau de sécurité de chaque élément logiciel. En rassemblant ces éléments logiciels, nous sommes en mesure de déterminer le niveau de sécurité de l'ensemble du système logiciel.
Figure 2 : Démontage et intégration du logiciel d'un dispositif médical
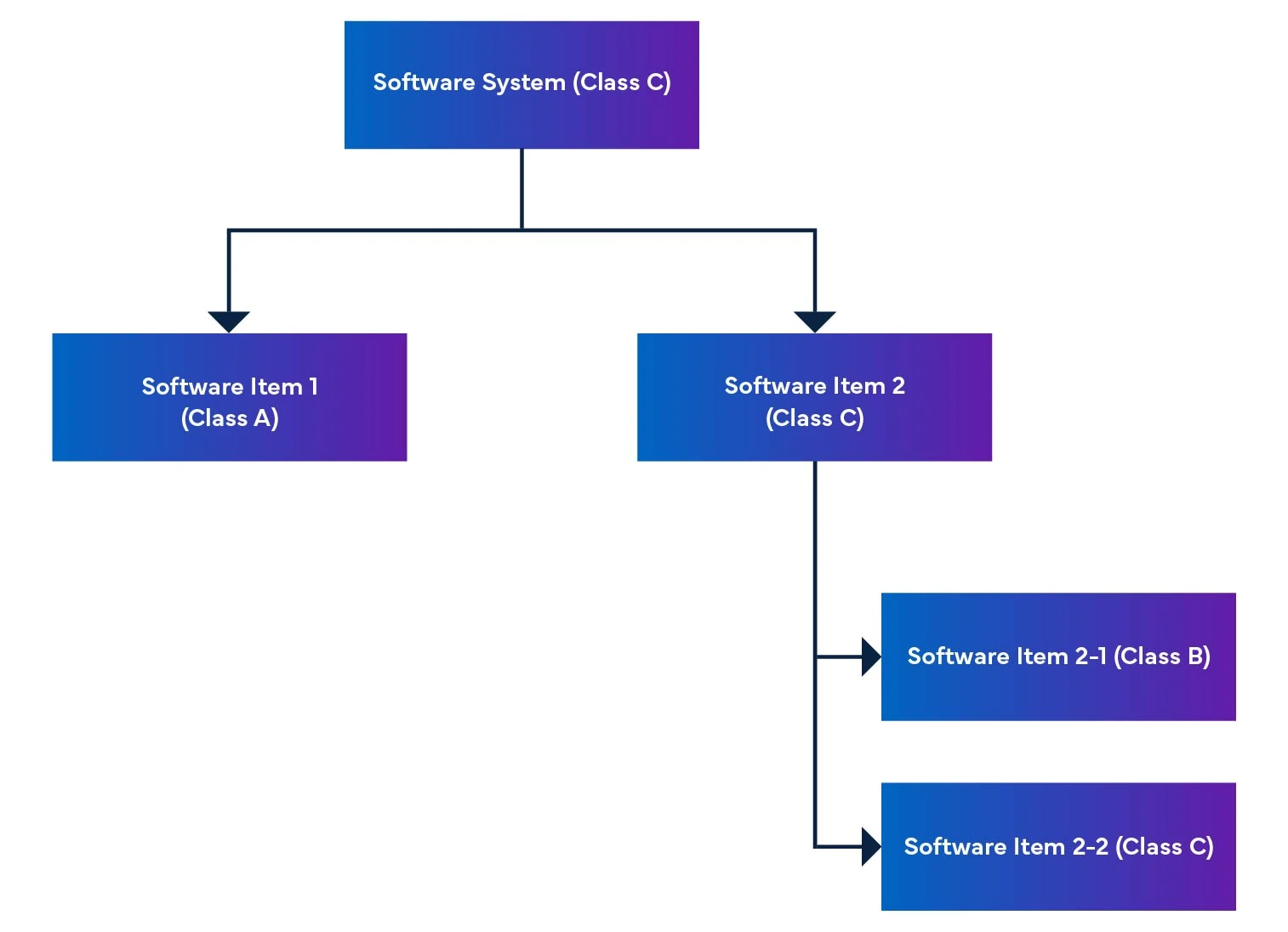
Le règlement mentionne également l'évaluation de la sécurité des logiciels, qui permet d'identifier les risques des logiciels SaMD (voir tableau 1).
Tableau 1 : Définition de l'évaluation de la sécurité
| Evaluation | Définition de la classe de sécurité logicielle des dispositifs médicaux |
| Classe A | Aucune possibilité de blessure ou de préjudice corporel. |
| Classe B | Des blessures moins graves (blessures mineures) sont probables. |
| Classe C | Risque de blessures graves ou mortelles. |
Gestion de la configuration des logiciels
- Maintenir une documentation précise et à jour pour toutes les versions, modifications et mises à jour des logiciels.
- Veiller à ce que tous les documents soient correctement examinés et approuvés.
- Mise en œuvre de procédures de gestion des changements de configuration des logiciels.
- Documenter toutes les activités et décisions relatives à la gestion de la configuration des logiciels.
Maintenance des logiciels
- tester et contrôler régulièrement le logiciel pour s'assurer qu'il reste sûr et efficace pour l'usage auquel il est destiné.
- Mettre en œuvre des procédures pour résoudre les problèmes éventuels, y compris les corrections de bogues et les mises à jour de logiciels.
- Documenter toutes les activités et décisions relatives à la maintenance des logiciels.
Dépannage
- Identifier la cause profonde du problème.
- Mettre en œuvre des actions correctives pour résoudre le problème.
- Documenter l'ensemble du processus de dépannage pour référence ultérieure.
En suivant les lignes directrices ci-dessus, les développeurs peuvent s'assurer que tous les problèmes liés à leur logiciel de dispositif médical sont correctement traités et documentés, et que le logiciel répond aux exigences nécessaires pour l'approbation ou l'examen.
Si vous êtes un fabricant de dispositifs médicaux souhaitant se conformer aux normes logicielles sud-coréennes en matière de dispositifs médicaux, les experts en réglementation de Freyrpeuvent vous guider dans le paysage réglementaire complexe du pays. Nous veillerons à ce que vos dispositifs soient conformes aux dernières réglementations sud-coréennes en matière de dispositifs médicaux afin d'assurer une conformité sans faille. Contactez-nous us en savoir plus !









