3 minutes de lecture
Au fil des ans, les progrès des logiciels et de la numérisation ont entraîné un changement radical dans la manière dont les dispositifs médicaux sont administrés et fournis. L'intégration des logiciels aux dispositifs médicaux s'est rapidement développée et a permis des avancées incroyables dans la fourniture de solutions de soins de santé dans divers domaines tels que le diagnostic, la prévention des maladies et le traitement d'une blessure ou d'une maladie.
Cependant, l'effet des logiciels sur la sécurité et les performances des dispositifs médicaux a été remis en question, en particulier lorsque le dispositif lui-même est un produit exclusivement logiciel. Par conséquent, la réglementation relative aux logiciels pour dispositifs médicaux est constamment révisée afin de déterminer si software as a medical device doivent être considérés software as a medical device SaMD). Récemment, le comité consultatif de la European Commission le Groupe de coordination des dispositifs médicaux (MDCG), s'est concentré sur l'amélioration de la réglementation relative aux logiciels des dispositifs médicaux et a publié un guide décrivant l'approche à adopter pour déterminer si un logiciel est un dispositif médical ou non. Que contient ce guide ? us .
Le champ d'application des orientations
Les orientations du MDCG couvrent à la fois les logiciels de dispositifs médicaux et les logiciels de dispositifs médicaux de diagnostic in vitro (DIV). Selon le document, un logiciel de dispositif médical (MDSW) est défini comme un logiciel destiné à être utilisé seul ou en combinaison, dans un but spécifié dans la définition d'un "dispositif médical" dans le règlement sur les dispositifs médicaux 2017/745 (MDR) ou le règlement sur les dispositifs médicaux de diagnostic in vitro 2017/746 (IVDR). Il décrit les critères à appliquer pour déterminer si un logiciel soumis à examen est un dispositif médical ou non et entend fournir des clarifications et des recommandations supplémentaires sur les FDSM pour les fabricants de dispositifs médicaux et les autres parties.
Tout d'abord, les orientations définissent les termes les plus importants utilisés dans le contexte de la DSMT, à savoir
Destination: l'utilisation pour laquelle un dispositif est prévu selon les données fournies par le fabricant sur l'étiquette, dans le mode d'emploi ou dans les documents ou déclarations promotionnels ou de vente, et comme spécifié par le fabricant dans l'évaluation clinique.
Accessoire: Un article qui, sans être lui-même un dispositif médical, est destiné par son fabricant à être utilisé avec un ou plusieurs dispositifs médicaux pour permettre spécifiquement l'utilisation du ou des dispositifs médicaux conformément à leur(s) destination(s) prévue(s) ou pour assister la fonctionnalité du ou des dispositifs médicaux spécifiquement et directement en termes de leur(s) destination(s) prévue(s). En outre, le MDCG mentionne que l'accessoire logiciel peut conduire ou influencer l'utilisation d'un dispositif médical et que les instructions d'utilisation et autres documents fournis par le fabricant doivent contenir des détails sur la manière dont le logiciel et les accessoires appropriés doivent être sélectionnés.
Logiciel : il s'agit d'un ensemble d'instructions qui traitent des données d'entrée et créent des données de sortie.
Détermination des logiciels de dispositifs médicaux
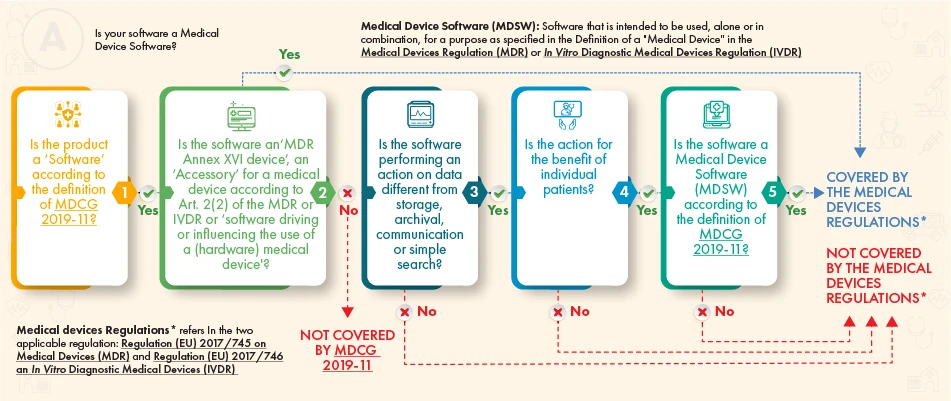
Selon l'organigramme ci-dessus, le logiciel en question devrait être soumis à la réglementation s'il répond aux critères suivants :
- la définition d'un dispositif médical ou d'un accessoire de celui-ci, ou la conduite des opérations du dispositif médical, ou
- Il effectue un traitement supplémentaire des données (pas seulement le stockage ou la communication) et son action crée des avantages pour les patients et répond à la définition d'un logiciel de dispositif médical conformément aux orientations du MDCG.
Détermination du logiciel pour les dispositifs médicaux de diagnostic in vitro
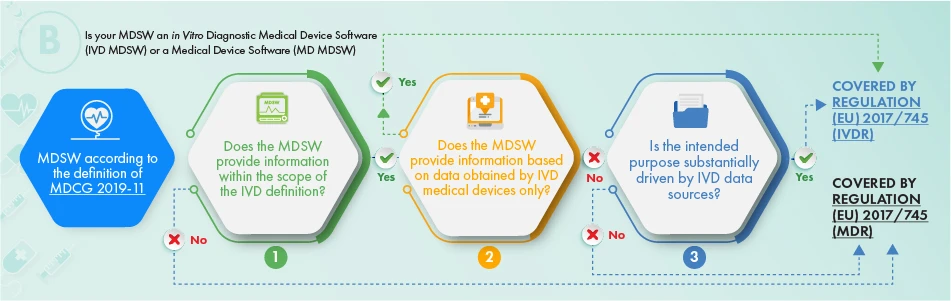
L'organigramme ci-dessus décrit l'approche à appliquer en ce qui concerne les produits destinés au diagnostic in vitro. Pour déterminer si le logiciel en question doit faire l'objet d'une réglementation, il convient d'examiner les critères suivants :
- la définition d'un dispositif médical ou d'un accessoire de celui-ci, ou la conduite des opérations du dispositif médical, ou
- Il fournit les informations habituellement fournies par les dispositifs médicaux de diagnostic in vitro et uniquement les informations recueillies à partir d'un dispositif médical de diagnostic in vitro, ou
- L'objectif du logiciel est de traiter les questions relatives à l'IVDR.
Selon les orientations du MDCG, le type d'interconnexion entre le logiciel de dispositif médical et le dispositif n'affecte pas la qualification du logiciel en tant que dispositif dans le cadre du MDR et de l'IVDR. Un logiciel de dispositif médical peut exister en tant que produit autonome ou être incorporé dans un dispositif matériel et clarifie les exigences réglementaires suivantes :
- Compte tenu de sa qualification et de sa classification, un logiciel de dispositif médical autonome doit être soumis à l'ensemble des procédures réglementaires conformément à la législation applicable.
- Un logiciel de dispositif médical qui fait partie intégrante d'un dispositif médical matériel pourrait être mis sur le marché dans le cadre de la procédure simplifiée. Il serait soumis à un examen non pas séparément, mais au cours de l'évaluation générale du dispositif médical matériel lui-même.
En résumé, les recommandations du MDCG couvrent les aspects essentiels liés à la classification des logiciels pour dispositifs médicaux et à la détermination des exigences réglementaires applicables. Les fabricants de dispositifs médicaux, les développeurs de logiciels et les autres parties concernées doivent suivre et mettre en œuvre les recommandations du MDCG afin de garantir leur conformité. Pour obtenir plus d'informations sur la classification software as a medical device votre software as a medical device, consultez un expert en réglementation. Restez informé. Restez en conformité.









