3 minutes de lecture
L'étiquetage fait partie intégrante de la commercialisation des dispositifs médicaux. L'étiquette est une information apposée sur le dispositif et/ou son emballage dans un format lisible par l'homme. L'étiquetage a pour principal objectif de fournir des informations de sécurité aux utilisateurs, qui peuvent être des professionnels de santé, des consommateurs ou toute autre personne concernée.
Toutes les autorités réglementaires mondiales ont certaines exigences en matière d'étiquetage. De même, l'UE a détaillé les exigences en matière d'étiquetage dans le chapitre III de l'annexe I du règlement européen sur les dispositifs médicaux (EU MDR) 2017/745. Il est avant tout important de noter que tous les symboles couvrant les informations requises doivent figurer sur l'étiquetage du dispositif et dans les documents (brochures, manuels, modes d'emploi, etc.) qui l'accompagnent.
Voici quelques-unes des considérations essentielles à prendre en compte en matière d'étiquetage pour se conformer au règlement EU MDR :
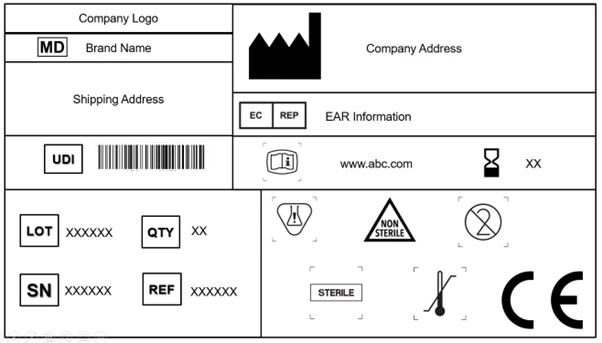
1. Symbologie de l'étiquetage des dispositifs médicaux
Chaque fabricant est tenu d'apposer le symbole du dispositif médical, qui indique que le produit fourni sur le marché de l'UE est un dispositif médical. Il est obligatoire d'apposer ce symbole sur le dispositif et à tous les niveaux de l'emballage. En outre, l'étiquette doit mentionner le nom commercial et le nom d'origine du dispositif.
2. Dispositifs spéciaux
Si le produit est un dispositif spécial ou personnalisé, son statut doit être mentionné sur l'étiquette. Par exemple, si le produit n'est destiné qu'à l'investigation clinique, l'étiquette doit le mentionner explicitement.
Pour les dispositifs contenant des matériaux absorbants ou susceptibles de se disperser localement dans le corps humain, l'étiquetage doit mentionner la composition du matériau et des détails quantitatifs sur les principaux constituants.
Un étiquetage explicite est même requis dans le cas des dispositifs à usage unique et des dispositifs stériles. Pour les dispositifs retraités, l'étiquetage doit mentionner le nombre de fois qu'ils peuvent être retraités, le nombre de fois qu'ils ont été retraités jusqu'à présent et la méthode de stérilisation utilisée.
3. Présence de substances toxiques
La déclaration de la présence de substances CMR (cancérogènes, mutagènes, toxiques pour la reproduction) et de perturbateurs endocriniens est obligatoire sur les étiquettes si la concentration est supérieure à 0,1 % en poids. La liste de ces substances doit être apposée sur le dispositif et/ou l'emballage.
En outre, un élément labelling présence un élément labelling dérivés sanguins et tissulaires (même lorsqu'ils sont contenus dans la substance médicinale du dispositif combiné) doit être apposé sur les dispositifs.
4. Normes harmonisées
Le règlement EU MDR reconnaît et accepte la norme ISO 15223-1: 2021. Ce document définit les symboles à utiliser pour l'étiquetage des dispositifs médicaux et de leur emballage. Le chapitre 3 (23.1, h) de l'annexe I du EU MDR que des symboles internationalement reconnus peuvent être utilisés et que, dans les régions où ces symboles ne sont pas reconnus, leur description doit être fournie dans un document accompagnant le dispositif.
5. UDI
Les articles 27, 28, 29 et l'annexe VI (A, B, C) détaillent les règles et réglementations relatives à l'UDI. L'étiquette doit désormais contenir un support UDI [identification automatisée pour la capture des données (AIDC) et représentation de l'UDI lisible par l'homme (HRI)] sur le dispositif et les niveaux d'emballage supérieurs. L'emballage supérieur du dispositif (à l'exclusion des emballages d'expédition) aura son propre support UDI.
6. Informations électroniques à utiliser (eIFU)
Les adresses web (URL) sous forme d'eIFU peuvent également être placées sur l'étiquetage du dispositif médical avec les IFU papier. Les eIFU peuvent être utilisés dans le cas de dispositifs médicaux implantables, implantables actifs, fixes et de logiciels (destinés également aux profanes).
7. Information des opérateurs économiques (OE)
L'étiquette contient généralement les informations du fabricant. Toutefois, dans le cas de fabricants étrangers, les informations du représentant autorisé doivent être placées sur les étiquettes commerciales.
8. Avertissements et précautions
Les avertissements et les précautions doivent être mentionnés sur l'étiquette du dispositif. Les informations sur cet aspect peuvent être réduites au minimum, et les détails peuvent être fournis sur l'IFU.
Les fabricants sont également tenus de se conformer aux exigences nationales en matière d'étiquetage. Les exigences linguistiques dépendent de l'État membre de l'UE. Elle peut avoir un impact considérable sur l'étiquetage, les notices d'utilisation et l'emballage du dispositif en termes de temps et de coûts.
Ces exigences supplémentaires peuvent encore alourdir le fardeau du fabricant en raison de la complexité du processus d'étiquetage existant. Un échec dans ce domaine peut s'avérer très coûteux, entraînant des rappels de produits et des mesures correctives et préventives (CAPA).
Vous avez besoin d'aide pour l'étiquetage conformément au règlement EU MDR? Freyr des services complets en matière d'étiquetage des dispositifs médicaux. N'hésitez pas à contacter dès maintenant nos experts en réglementation à l'adresse suivante : sales@freyrsolutions.com.









