
3 minutes de lecture
Le règlement européen sur les dispositifs médicaux (MDR) fait parler de lui depuis un certain temps déjà. Le MDR a remplacé la directive relative aux dispositifs médicaux (MDD) et la directive relative aux dispositifs implantables actifs (AIMDD). Au départ, la transition devait être entièrement effective en mai 2020, mais en raison de l'émergence de la pandémie de COVID-19, la mise en œuvre a été repoussée au 26 mai 2021. Dans ce calendrier, d'ici le 26 mai 2024, tous les certificats MDD deviendront caducs et les fabricants de dispositifs devront se conformer au EU MDR. En outre, les dispositifs MDD légalement mis sur le marché conformément aux directives 90/385/CEE et 93/42/CEE avant le 26 mai 2020, ainsi que les dispositifs mis sur le marché à partir du 26 mai 2020 en vertu d'un certificat, continueront d'être disponibles sur le marché jusqu'au 27 mai 2025. Les délais sont indiqués ci-dessous :
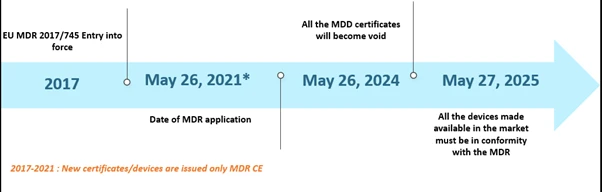
Chronologie des scénarios EU MDR de EU MDR
Cependant,la capacité limitée des organismes notifiés (ON) et le manque de préparation des fabricants ont posé certains défis dans la mise en œuvre du RDM dans les délais impartis. En octobre 2022, on comptait au total trente-huit (38) organismes notifiés (ON), qui ont reçu environ 8 120 demandes de EU MDR , parmi lesquelles 1 990 certificats ont été délivrés. Selon leurs estimations initiales, seuls 7 000 certificats pouvaient être traités, ce qui a conduit à une prolongation du calendrier. En outre, l'une des autres raisons probables de cette prolongation était de garantir la disponibilité continue des dispositifs médicaux sûrs dont les certificats ont déjà expiré ou expireront avant le 26 mai 2024. Le scénario actuel pour le calendrier prolongé est présenté ci-dessous.
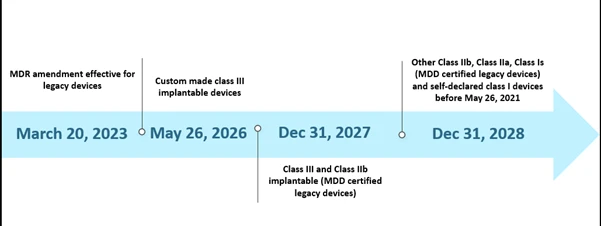
Chronologie des scénarios EU MDR de EU MDR
La nouvelle extension s'applique aux dispositifs anciens qui satisfont à l'article 120 (3e) et qui bénéficient d'un marquage CE ou d'une dérogation MDD valide au 20 mars 2023, et qui resteront sur le marché avec les dispositifs portant le marquage CE MDR. D'ici le 26 mai 2024, les fabricants de dispositifs anciens doivent avoir mis en place un système de gestion de la qualité et déposé une demande d'évaluation de la conformité auprès d'un ON désigné par le RIM, et d'ici le 26 septembre 2024, les fabricants de dispositifs anciens doivent avoir conclu un accord avec un ON désigné par le RIM.
Examinons maintenant l'impact que cette extension pourrait avoir sur les fabricants.
Possibilités offertes aux fabricants par cette extension :
- Accès élargi au marché pour les fabricants de dispositifs certifiés MDD/AIMDD qui ont déjà pris des mesures pour se conformer au RDM.
- Les fabricants certifiés MDR dont les certificats MDD/AIMDD CE n'ont pas été révoqués sont autorisés à mettre sur le marché des dispositifs anciens jusqu'à la fin de la période de transition, en plus de leurs dispositifs conformes au MDR.
- Les fabricants qui bénéficient d'une dérogation nationale au 20 mars 2023 peuvent bénéficier de la période transitoire.
- La période de prolongation donne plus de temps pour mieux comprendre les règles et les règlements, ce qui permet de rationaliser le processus et de se conformer au RIM.
Défis que pourraient rencontrer les fabricants avec cette extension :
- Il n'y a aucun avantage commercial pour les fabricants d'appareils traditionnels qui ne souhaitaient pas se conformer au règlement MDR.
- L'extension du MDR peut faire traîner les processus de certification et retarder le lancement des produits, ce qui est une conséquence directe de l'arriéré des examens effectués par les ON.
Quelles mesures les fabricants doivent-ils prendre ?
- Il est impératif que les fabricants déterminent la classe de risque MDR de leur dispositif médical afin d'identifier rapidement le délai de transition approprié conformément aux réglementations MDR modifiées.
- Pour garantir la conformité avec les réglementations du RIM, il est essentiel d'identifier et d'entamer une communication avec les ON désignés par le RIM qui possèdent les compétences spécifiques requises pour la classification de votre dispositif médical.
- Il est essentiel de procéder à une évaluation complète des lacunes de votre dispositif médical certifié selon la directive MDD/AIMDD, d'identifier et de traiter toute non-conformité avec les réglementations MDR et de garantir une mise en conformité dans les délais impartis.
Il est essentiel que les fabricants prennent immédiatement des mesures pour se conformer au RDM. Le délai prolongé offre aux fabricants certaines opportunités pour se conformer au RDM, mais il présente également des défis, tels que le retard des processus de certification et le coût de la mise en conformité. Pour relever ces défis et tirer parti des opportunités, laissez notre équipe de professionnels vous aider tout au long du processus de mise en conformité au RDM et vous assurer le succès dans cet environnement réglementaire difficile. Prenez rendez-vous avec us pour en savoir plus sur la manière dont nous pouvons vous aider à vous conformer au RDM et à garder une longueur d'avance. Restez informé. Restez conforme.









