
3 minutes de lecture
Le formulaire 510(k) est une demande préalable à la mise sur le marché adressée à la FDA démontrer que le dispositif destiné à être commercialisé est aussi sûr et efficace, c'est-à-dire substantiellement équivalent, qu'un dispositif déjà commercialisé légalement (prédicat). Les dispositifs présentant un risque modéré doivent faire l'objet d'une notification 510(k), ce qui concerne une minorité de dispositifs de classe I et III et une majorité de dispositifs de classe II.
Il existe trois (03) types de programmes 510(k) : traditionnel, abrégé et spécial. La voie de la sécurité et de la performance a été introduite en 2019 et s'appuie sur le programme abrégé. Le programme eSTAR, introduit en 2020, permet de soumettre un dispositif médical complet au moyen d'un formulaire PDF interactif.
Qui a besoin d'une certification 510(k) ?
Le 510(k) est essentiellement le nom du processus/parcours que US les fabricants de dispositifs médicaux souhaitant commercialiser leurs dispositifs à risque modéré à élevé aux US afin de démontrer que le produit à commercialiser est aussi sûr et efficace qu'un dispositif commercialisé légalement.
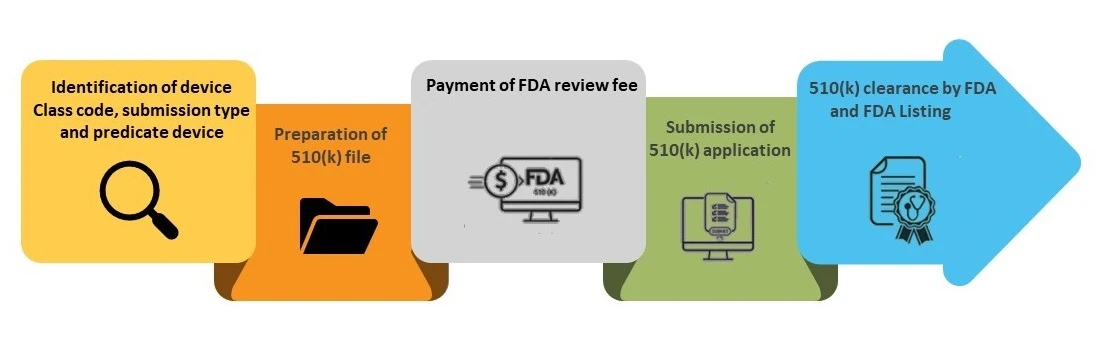
La procédure d'obtention d'une autorisation 510(k) est décrite ci-dessous, étape par étape.
Étape 1 - Identification du code de classe de l'appareil, du type de soumission et de l'appareil prédicat
- Identifier le code produit et le numéro de réglementation - Pour déterminer les exigences du test 510(k), il est nécessaire d'identifier au préalable le code produit et le numéro de réglementation. Il est possible d'effectuer une recherche dans la FDA afin de trouver le numéro de réglementation à 7 chiffres correspondant à l'utilisation prévue du dispositif en question.
- Le code FDA se compose de trois (03) lettres. Ce code permet de trouver des informations concernant la classification du produit, la description de la réglementation et les exigences en matière de bonnes pratiques de fabrication (BPF).
- Sélection du type de demande - Le demandeur peut choisir l'un des trois (03) types de demande mentionnés précédemment. Le formulaire 510(k) traditionnel est destiné aux premières soumissions, le formulaire 510(k) spécial est destiné aux fabricants de dispositifs médicaux qui souhaitent soumettre des modifications à un dispositif existant et le formulaire 510(k) abrégé peut être choisi lorsque le dispositif est conforme aux normes consensuelles volontaires établies. Dans le cas d'un formulaire 510(k) abrégé, le demandeur doit se référer aux documents FDA .
- Identification du dispositif prédicat - Un fabricant de dispositifs médicaux doit prouver que le dispositif qu'il a l'intention de commercialiser a la même utilisation prévue et les mêmes caractéristiques techniques que le dispositif légalement commercialisé, également connu sous le nom de dispositif prédicat. S'il existe des différences dans les caractéristiques techniques, le demandeur doit prouver qu'aucun problème de sécurité et d'efficacité n'est associé à ces différences.
Étape 2 - Préparation du dossier 510(k)
L'étape suivante consiste à préparer le dossier 510(k), les directives et les informations, qui sont disponibles sur le FDA . Il comprend la liste de contrôle d'acceptation pour les trois (03) types de programmes 510(k) et un microsite intitulé « Content for 510(k) », qui contient des informations concernant les déclarations relatives aux indications d'utilisation, la comparaison d'équivalence substantielle et l'étiquetage proposé, entre autres informations utiles.
Étapes de la procédure de soumission 510(k)
Étape 3 - Paiement des frais FDA
Tous les types de demandes 510(k) sont soumis à la redevance d'utilisation. Pour l'exercice 2023, la redevance standard pour les 510(k) est de 19 870 $. Pour les entreprises certifiées par le Centre for Diagnostics and Radiological Health (CDRH), également connues sous le nom de petites entreprises, la redevance est de 4 967 $. La redevance est susceptible d'être modifiée au cours du prochain exercice financier.
Étape 4 - Soumission de la demande 510(k)
Le soumissionnaire peut envoyer une copie électronique (eCopy) ou un modèle de soumission et une ressource électroniques (eSTAR) pour une soumission de précommercialisation via le portail du CDRH.
À partir du 1er octobre 2023, toutes les demandes 510(k), à moins qu'elles ne soient exemptées conformément à la directive finale, devront être soumises par voie électronique à l'aide d'eSTAR.
Une fois le formulaire 510(k) soumis, un numéro de contrôle unique est attribué, appelé « numéro 510(k) » ou « numéro K ». FDA deux vérifications : la première pour s'assurer que les frais d'utilisation ont bien été payés, et la seconde pour vérifier qu'une copie électronique ou un eSTAR valide a bien été fourni.
- Au septième jour, FDA une lettre d'accusé de réception si les frais d'utilisation ont été acquittés et qu'une copie électronique ou un eSTAR valide a été fourni. Dans le cas contraire, la FDA une lettre de suspension pour les questions non résolues.
- Au 15e jour, FDA un examen d'acceptation. FDA le demandeur si la demande 510(k) est acceptée pour examen approfondi ou placée en attente de refus d'acceptation (RTA).
- Au 60e jour, FDA un examen approfondi. FDA via une interaction approfondie d'
s pour informer que FDA à un examen interactif ou que la demande 510(k) sera mise en attente et que des informations supplémentaires seront demandées.
Étape 5 - FDA et inscription dans la base de données FDA (k)
L'objectif de la FDA d'annoncer sa décision relative aux amendements concernant les frais d'utilisation des dispositifs médicaux (MDUFA) dans un délai de 90 FDA . FDA correspondent aux jours calendaires entre la date de réception du formulaire 510(k) et la date de la décision MDUFA, à l'exclusion des jours pendant lesquels la demande a été suspendue pour obtenir des informations supplémentaires. Les décisions MDUFA pour les demandes 510(k) comprennent les conclusions de « substantiellement équivalent » (SE) ou « non substantiellement équivalent » (NSE).
Une fois la décision prise, FDA une lettre de décision au demandeur par courrier électronique. Une demande 510(k) qui reçoit une lettre de décision SE est considérée comme « approuvée ». Elle est ensuite répertoriée dans la base de données 510(k) avec les indications d'utilisation du dispositif médical et le résumé 510(k) ou la déclaration 510(k) en pièces jointes.
On peut en conclure qu'une planification et une exécution minutieuses, s'appuyant sur une documentation exhaustive et une compréhension approfondie de l'environnement réglementaire, sont essentielles pour réussir une demande 510(k) auprès de FDA.
Pour obtenir de l'aide concernant le processus de soumission 510(k) de votre dispositif médical, vous pouvez us écrire us l'adresse sales@freyrsoltions.com ou prendre rendez-vous avec nos experts, qui vous aideront à vous y retrouver dans les procédures. Restez informé. Restez en conformité.









