3 min read
wyroby medyczne Unii Europejskiej w wyroby medyczne (EU MDR) 2017/745 definiuje zakres wyroby medyczne sterylnych wyroby medyczne wyroby, które muszą być wolne od mikroorganizmów (bakterii, wirusów itp.). Wyroby klasy I o funkcji sterylnej mogą również uzyskać certyfikat własny, ale różni się on pod względem wymagań w porównaniu z innymi podkategoriami wyrobów klasy I.
Wymagania dotyczące wyrobów klasy I różnią się znacznie w zależności od etapu produkcji do wprowadzenia wyrobu na rynek. To samo dotyczy wyroby medyczne klasy I wyroby medyczne warunkach sterylnych. Na przykład, jeśli jest to wyrób klasy I o funkcji sterylnej, wymagania dotyczące produkcji i ustanowienia Quality Management System (QMS) znacznie różnić w porównaniu z wyrobami klasy I o funkcjach pomiarowych.
Niniejszy artykuł ma na celu przedstawienie zwięzłej listy kontrolnej, którą należy wziąć pod uwagę przy wprowadzaniu do obrotu w Unii Europejskiej wyrobu klasy I w warunkach sterylnych. Lista kontrolna pomoże producentom w podejmowaniu świadomych decyzji dotyczących różnych aspektów.
Dos!
Znajomość zakresu przed rozpoczęciem produkcji
Zgodnie z rozporządzeniem EU MDR wyroby medyczne funkcjach sterylnych muszą być opracowywane i produkowane przy użyciu odpowiednich procedur zapewniających sterylność do momentu wprowadzenia ich do obrotu. Urządzenia mogą być ponownie użyte dopiero po odpowiednim czyszczeniu, dezynfekcji i sterylizacji.
Wybór właściwych praktyk sterylizacji
Producenci mają do wyboru różne metody sterylizacji urządzeń. Metoda tlenku etylenu (EtO) jest szeroko stosowana do sterylizacji wyroby medyczne. Wybór metody sterylizacji musi być odpowiednio uzasadniony. Niektóre normy, które producenci mogą stosować w celu ustanowienia, zapewnienia i utrzymania sterylizacji to EN ISO 14937, ISO TS 21387, ISO TS 13004, ISO 11137 i inne.
Należy również pamiętać, że dokumentacja szczegółowej metody sterylizacji, metody stosowanej w trakcie procesu oraz walidacji danych musi być sporządzona dokładnie. Informacje dotyczące sterylizacji, takie jak temperatura ogrzewania, warunki panujące w pomieszczeniu, walidacje procesu itp., muszą być dostarczone zgodnie z EU MDR .
Znajomość wymogów i wytycznych, których należy przestrzegać
Jednym z kluczowych kroków jest udokumentowanie wymagań technicznych i zapoznanie się z normami przyjętymi w ramach EU MDR. Na przykład norma EN 556 jest europejską normą dotyczącą urządzeń sterylizowanych. Norma EN 556 szczegółowo określa wymagania dotyczące oznaczania wyrobów medycznych jako sterylne. Ponadto norma ISO TS 19930 określa strategie zapewniania sterylności urządzeń. Nie została ona jednak jeszcze przyjęta jako norma unijna.
Poznaj swoją trasę
W celu uzyskania znaku CE należy zapoznać się z udziałem jednostek notyfikowanych oraz ścieżką oceny zgodności, którą należy podjąć. Ponadto wyroby medyczne warunkach sterylnych muszą być zgodne z załącznikami IX i XI.
Zaangażowanie jednostki notyfikowanej jest wymagane, ale ograniczone. W przypadku wyrobów sterylnych zaangażowanie jednostek notyfikowanych będzie dotyczyło utrzymania i zabezpieczenia warunków sterylności. Na przykład oceny zgodności będą obejmować oceny mikrobiologiczne, metody aseptyczne itp.
Zakazy!
Nie przegap wymogów dotyczących pakowania i etykietowania

W przypadku wyrobów sterylnych obowiązują dodatkowe wymogi dotyczące zgodności opakowań i etykiet. Spełniając wymagania dotyczące pakowania i etykietowania, nie należy zapominać o informacjach, które należy umieścić. Pierwszym i najważniejszym krokiem jest umieszczenie symbolu sterylności (wspomnianego poniżej) na etykiecie urządzenia.
Należy podać takie informacje, jak deklaracja sterylności, metoda sterylizacji, instrukcje na wypadek uszkodzenia lub niezamierzonego otwarcia opakowania sterylnego itp. Należy również podać okres trwałości produktu sterylnego i proces ponownej sterylizacji (jeśli dotyczy). Na wyrobie należy również umieścić numer identyfikacyjny jednostki notyfikowanej.
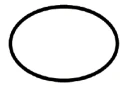
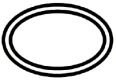
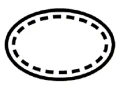
Niektóre symbole, które należy dodać (w zależności od zastosowania) na etykietach sterylnych wyroby medyczne :
Symbol | Wskazanie |
| Sterylne wyroby medyczne sterylizacją metodą stosowaną jako technika aseptyczna |
| Sterylne wyroby medyczne zastosowano metodę sterylizacji tlenkiem etylenu |
| Sterylne wyroby medyczne zastosowano metodę sterylizacji suchym ciepłem lub parą wodną |
| Sterylne wyroby medyczne sterylizacją metodą pary wodoru |
| Sterylne wyroby medyczne sterylizacją metodą promieniowania |
| Urządzenie nie może zostać ponownie wysterylizowane |
| Urządzenie nie jest sterylizowane |
| Nie używać, jeśli opakowanie jest uszkodzone |
| System pojedynczej bariery sterylnej |
| System podwójnej bariery sterylnej |
| Pojedynczy sterylny system barierowy z wewnętrznym opakowaniem ochronnym |
| Pojedynczy system bariery sterylnej z zewnętrznym opakowaniem ochronnym |


Nie zapomnij o wymaganiach dotyczących umieszczenia po wprowadzeniu na rynek
Wyroby sterylne są zobowiązane do prowadzenia rejestru zdarzeń niepożądanych w przypadku ich wystąpienia. W takim przypadku zarejestrowane zdarzenia, takie jak dokumenty, muszą zostać przedstawione na żądanie właściwych organów.
Rozporządzenie EU MDR szczegółowe wymagania dotyczące samocertyfikacji. Jest to złożona procedura, której głównym czynnikiem są różne klasy w ramach wyrobów klasy I oraz różne aspekty wymagań dotyczących uzyskania znaku CE. Producenci wyrobów klasy I przeznaczonych do stosowania w warunkach sterylnych powinni być świadomi zakresu i szczegółowych wymagań dotyczących oznaczania.
Chcesz szczegółowo poznać różne aspekty urządzeń sterylnych klasy I? Chcesz omówić tę kwestię z doświadczonym ekspertem? Reach z us.









