
2 min read
US FDA wytyczne, które pomogą branży i pracownikom Agencji Zdrowia (HA) określić, kiedy zmiana oprogramowania urządzenia medycznego wymaga od producenta złożenia i uzyskania FDA na nowe zgłoszenie przed wprowadzeniem do obrotu (510(k)). Wytyczne te mają na celu zwiększenie przewidywalności, spójności i przejrzystości procesu podejmowania decyzji dotyczących terminu złożenia wniosku poprzez przedstawienie najmniej uciążliwego podejścia oraz opisanie ram regulacyjnych, polityk i praktyk leżących u podstaw takiej decyzji, w szczególności w odniesieniu do zmian oprogramowania. Przyjrzyjmy się szczegółowo FDA .
FDA i schemat blokowy
Aby pomóc producentom urządzeń medycznych w stosowaniu głównych zasad, dokument zawiera schemat blokowy, dodatkowe wyjaśnienia i przykłady potrzebne do podjęcia decyzji dotyczących nowego zgłoszenia przed wprowadzeniem do obrotu 510(k) w przypadku zmiany oprogramowania w urządzeniu już zatwierdzonym w US. Ponadto, korzystając z niniejszych wytycznych w celu ustalenia, czy należy złożyć nowe zgłoszenie 510(k) w celu zmiany istniejącego urządzenia, należy przestrzegać kilku zasad przewodnich. Niektóre z nich są powszechnie znane i wynikają z aktualnej polityki FDA (k), a inne są niezbędne do stosowania schematu logicznego wspomnianego w niniejszych wytycznych. Zgodnie z wytycznymi, przedstawiony schemat nie może obejmować wszystkich możliwych zawiłości związanych z takimi zmianami i ich wpływem na decyzję. Dlatego też, aby ustalić konieczność złożenia nowego zgłoszenia przed wprowadzeniem do obrotu 510(k), producenci wyrobów medycznych powinni wziąć pod uwagę ogólne zasady i schemat blokowy podsumowane poniżej.
- Zmiany wpływające na bezpieczeństwo lub skuteczność urządzenia
- Wstępna ocena ryzyka
- Niezamierzone konsekwencje zmian
- Wykorzystanie zarządzania ryzykiem
- Rola testowania (weryfikacji i walidacji) w ocenie, czy zmiana może znacząco wpłynąć na bezpieczeństwo i skuteczność.
- Ocena jednoczesnych zmian w celu ustalenia, czy wymagane jest złożenie nowego 510(k).
- Odpowiednie urządzenie porównawcze i skumulowany efekt zmian
- Wymóg dotyczący dokumentów (21 CFR część 820)
- Zgłoszenia 510(k) dla zmodyfikowanych urządzeń
- Ustalenia dotyczące istotnej równoważności
- Złożenie nowego 510(k) jest prawdopodobnie wymagane, jeśli producent zmodyfikuje swoje urządzenie, aby wpłynąć na jego bezpieczeństwo lub skuteczność. Jednak zmiany, które nie mają na celu wpłynięcia na bezpieczeństwo lub skuteczność urządzenia, powinny nadal podlegać ocenie.
- Aby ustalić, czy zmiana lub modyfikacja może znacząco wpłynąć na bezpieczeństwo lub skuteczność, producent powinien najpierw przeprowadzić ocenę opartą na ryzyku, aby dowiedzieć się, czy zmiana może mieć pozytywny lub negatywny wpływ na bezpieczeństwo lub skuteczność wyrobu. Ta ocena ryzyka powinna identyfikować i analizować wszystkie nowe zagrożenia i zmiany w istniejących zagrożeniach wynikających ze zmiany wyrobu i prowadzić do wstępnej decyzji, czy wymagane jest złożenie nowego zgłoszenia 510(k).
- Czasami istnieją dodatkowe niezamierzone lub nieplanowane konsekwencje, które mogą zostać wywołane podczas składania oprogramowania. Schemat blokowy powinien ocenić te konsekwencje, aby określić, czy konieczne jest złożenie nowego 510(k).
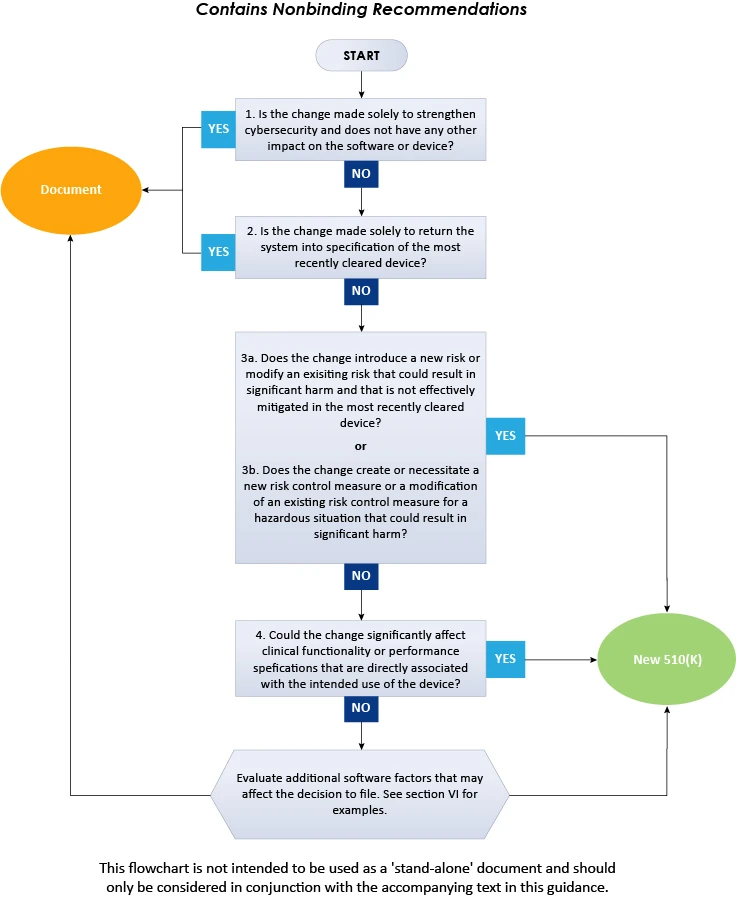
Powyższy schemat blokowy ilustruje procedurę krok po kroku, którą należy zastosować przy podejmowaniu decyzji o złożeniu wniosku 510(k) w przypadku zmian oprogramowania w istniejących urządzeniach. Podsumowując, obecne FDA szczegółowo opisują podejście, które powinni stosować producenci urządzeń medycznych przy podejmowaniu decyzji, czy zmiany oprogramowania w istniejącym urządzeniu medycznym wymagają złożenia nowego wniosku 510(k). Aby uzyskać więcej informacji na temat FDA , skonsultuj się Freyr – sprawdzonym ekspertem w dziedzinie regulacji prawnych. Bądź na bieżąco. Zachowaj zgodność z przepisami.









