5 min read
Nadsyłanie zgłoszeń zaplanowano na 24 września 2016 r.
Po wdrożeniu drugiej fazy zgodności z UDI dla wyroby medyczne klasy III I/LS/LS wielu producentów urządzeń, zwłaszcza klasy II, zastanawia się, jak najlepiej przygotować się do terminu składania danych dotyczących urządzeń klasy II, który upływa 24 września 2016 r. Aby dać im wskazówki, w firmie Freyr zidentyfikowaliśmy kilka warunków wstępnych, które powinni wziąć pod uwagę, aby dostosować urządzenia klasy II do wymogów FDAdotyczących identyfikacji urządzeń medycznych (UDI).
Nowe przepisy wymagają, wyroby medyczne wszystkie wyroby medyczne klasy II były oznakowane i pakowane z unikalnym identyfikatorem urządzenia (UDI) oraz wprowadzone do globalnej bazy danych unikalnych identyfikatorów urządzeń (GUDID) FDA. Biorąc pod uwagę zmienność wymagań dotyczących zgodności w połączeniu z krótszymi terminami składania wniosków, wyzwaniem dla producentów urządzeń jest zapoznanie się ze szczegółami procesów zapewniania zgodności. Jednocześnie muszą oni zapewnić, że żadna z kluczowych cech urządzenia nie zostanie pominięta podczas gromadzenia rozproszonych danych dotyczących urządzeń w różnych systemach i uzgadniania ich w arkuszach kalkulacyjnych w celu tworzenia raportów zgodności.
Aby pomóc producentom urządzeń w łatwym przejściu przez ten czasochłonny i złożony proces zapewnienia zgodności, bez popełnienia błędów, Freyr poniższe wymagania wstępne, które należy spełnić.
Określ datę zgodności z UDI: Od momentu FDA ostatecznej decyzji niektóre daty zgodności urządzeń uległy zmianie i FDA przedłużone. Aby skrupulatnie zaplanować strategie i procesy związane z zapewnieniem zgodności oraz uniknąć pośpiesznych zmian w ostatniej chwili, producenci etykiet powinni określić dokładną datę zgodności.
Data zgodności urządzeń klasy II Wymagania dotyczące zgodności 24 września 2016 r.Wyroby klasy III, które muszą być oznakowane kodem UDI, muszą posiadać kod UDI jako trwałe oznakowanie na samym wyrobie, jeśli jest to wyrób przeznaczony do wielokrotnego użytku i przeznaczony do ponownego przetworzenia przed każdym użyciem. Etykiety i opakowania wyroby medyczne klasy IImuszą być opatrzone identyfikatorem UDI. Daty na etykietach tych urządzeń muszą być sformatowane zgodnie z wymaganiami Samodzielne oprogramowanie klasy II musi zgodnie z wymaganiami dostarczać swój kod UDI Dane dotyczące urządzeń klasy II, które muszą być oznakowane kodem UDI, muszą być przekazywane do bazy danych GUDID. W przypadku większości urządzeń data zgodności dla bezpośredniego znakowania jest inna niż w przypadku pozostałych wymogów. W oparciu o kategorię produktu, przeznaczoną do ponownego użycia lub do ponownego przetworzenia, należy określić datę zgodności z kodem UDI dla oznakowania bezpośredniego, jak pokazano tutaj:
Daty zgodności z oznaczeniami bezpośrednimi Kategoria urządzenia - ponownie użyte i przetworzone 24 września 2015 r. Urządzenia podtrzymujące i podtrzymujące życie, niezależnie od klasy urządzenia 24 września 2016 r. Urządzenia klasy III i urządzenia licencjonowane na mocy ustawy o publicznej służbie zdrowia 24 września 2018 r. Urządzenia klasy II 24 września 2020 r. Urządzenia klasy I i urządzenia niesklasyfikowane Ocena konieczności bezpośredniego oznaczania numeru UDI: Wszystkie wyroby medyczne są używane więcej niż jeden raz lub które muszą być ponownie przetwarzane przed każdym użyciem, muszą mieć bezpośrednie oznaczenie UDI. Wyjątek stanowią wyroby wszczepialne, które nie wymagają bezpośredniego oznaczania zgodnie z zasadą UDI. Urządzenia jednorazowego użytku, nawet jeśli są ponownie przetwarzane, również nie muszą być opatrzone trwałym oznaczeniem UDI – 21 CFR 801.45(d)(3). W związku z tym należy ocenić potrzebę bezpośredniego znakowania w oparciu o kategorię wyroby medyczne .
- Plan kompleksowej zgodności: Zapoznaj się z FDA dotyczącymi konkretnych produktów, które mają być zgodne z przepisami. Przeprowadź dokładną analizę luk, aby zidentyfikować braki związane z danymi lub technologią, które mogą utrudniać spełnienie rygorystycznych FDA . Niektóre z tych wyzwań mogą obejmować uzyskanie informacji DI lub PI oraz obsługę dużych ilości nieustrukturyzowanych danych pochodzących z różnych źródeł itp. Zamiast pracować do późna w nocy, aby zsynchronizować wszystkie dane dotyczące urządzeń medycznych w ostatniej chwili, należy z wyprzedzeniem zaplanować kompleksową zgodność z przepisami za pomocą sprawdzonych systemów i narzędzi, które wspierają integrację danych, jakość danych i zarządzanie danymi.
![]()
Uzyskaj numer DI i członkostwo w agencji: UDI składa się z identyfikatora urządzenia (DI – unikalny numer oparty na wersji lub modelu urządzenia) oraz identyfikatora produktu (PI – zawiera numer partii, numer seryjny lub datę ważności). Część DI identyfikatora UDI będzie służyć jako główny klucz do wyszukiwania informacji o urządzeniu w bazie GUDID. W celu przypisania identyfikatora DI FDA trzy agencje wydające identyfikatory – GS1, HIBCC i ICCBBA. W tym scenariuszu producenci etykiet muszą uzyskać członkostwo w jednej z agencji, aby otrzymać numer DI, który należy wprowadzić do bazy GUDID FDA.
![]()
Identyfikator Atrybuty Agencje wydające UDI DI (identyfikator urządzenia - dane statyczne)
Wymagana synchronizacja z GUDIDUnikalna liczba
Producent
Marka urządzenia
Model urządzenia
GSI
HIBCC
ICCBBAPI (Identyfikator produktu - dane dynamiczne)
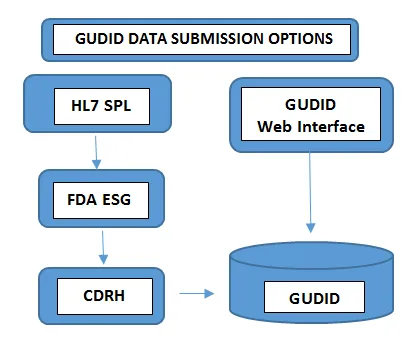
Wymagane na wszystkich poziomach opakowania.Numer partii, numer seryjny, data produkcji, data ważności, - Prześlij daneSposób przesyłania danych do GUDID zależy od wielkości portfolio produktów, którymi się zajmujesz. Producenci urządzeń z minimalną liczbą urządzeń decydują się na ręczne przesyłanie informacji UDI za pośrednictwem bezpłatnego interfejsu internetowego GUDID FDA. W takim przypadku za pośrednictwem zabezpieczonego interfejsu internetowego GUDID można przesłać tylko jeden rekord DI na raz.W innym przypadku producenci posiadający większą liczbę produktów wybierają opcję przesyłania HL7 SPL, aby zebrać dane elektronicznie i przekonwertować skonsolidowane dane do formatu SPL przed przesłaniem ich do elektronicznej bramki przesyłania FDA(ESG) FDA, używając numeru DUNS. Należy pamiętać, że konto GUDID nie jest związane z typem przesyłania danych. Konto służy do identyfikacji użytkownika, czyli podmiotu oznaczającego produkt, aby umożliwić przesyłanie danych dotyczących urządzeń za pomocą obu opcji.
![]()
- Załóż konto GUDID: Producent etykiet / urządzeń potrzebuje jednego lub więcej kont GUDID w zależności od liczby przydzielonych ról, takich jak koordynator GUDID, użytkownik wprowadzający dane itp. Jednak aby nadać każdej roli uprawnienia do wprowadzania danych, producent musi uzyskać zgodę FDA utworzeniem konta. Proces tworzenia odpowiedniego konta GUDID obejmuje wysłanie wniosku pocztą elektroniczną do FDA czym wnioskodawca, czyli Ty, otrzyma dokument wniosku o utworzenie konta do wypełnienia. Po odesłaniu wypełnionego dokumentu do FDA agencja sprawdzi formularz, a następnie wyśle wiadomość e-mail z danymi logowania do konta GUDID.


Wdrożenie UDI to złożony i czasochłonny proces. Podczas tego procesu, spełniając wymagania FDAdotyczące UDI, producenci urządzeń medycznych napotykają wiele wyzwań związanych z zarządzaniem danymi, integracją danych i przekazywaniem danych. Ponieważ termin dostosowania się do wymogów dla urządzeń klasy II upływa za rok, Freyr przedsiębiorstwom rozpoczęcie prac już teraz.
Aby pomóc Twojej organizacji przejść przez ten złożony proces zapewnienia zgodności, Freyr najlepsze z obu światów – w pełni konfigurowalne oprogramowanie UDI dostępne na żądanie,Freyr , a także Centrum Doskonałości (CoE), które oferuje najlepsze w swojej klasie, opłacalne i dostosowane do indywidualnych potrzebusługi UDI, oparte na Twoich unikalnych i wymagających wymaganiach.









