3 min read
Z biegiem lat, wraz z postępem w dziedzinie oprogramowania i cyfryzacji, nastąpiła ogromna zmiana w sposobie wyroby medyczne i dostarczania wyroby medyczne . Integracja oprogramowania z wyroby medyczne gwałtownie wyroby medyczne i przyczyniła się do niesamowitego postępu w dostarczaniu rozwiązań medycznych w różnych dziedzinach, takich jak diagnostyka, profilaktyka chorób oraz leczenie urazów i schorzeń.
Jednak wpływ oprogramowania na bezpieczeństwo i działanie wyroby medyczne niepewny, zwłaszcza gdy samo urządzenie jest produktem wyłącznie oprogramowaniem. W związku z tym przepisy dotyczące oprogramowania wyrobów medycznych są stale aktualizowane w celu określenia, czy oprogramowanie należy uznać za wyrób medyczny (SaMD). Niedawno rada doradcza European Commission Grupa Koordynacyjna ds. Wyrobów Medycznych (MDCG) – skoncentrowała się na ulepszeniu przepisów dotyczących oprogramowania wyrobów medycznych i opublikowała wytyczne opisujące podejście, które należy stosować przy ustalaniu, czy oprogramowanie jest wyrobem medycznym, czy nie. Co zawierają te wytyczne? Przyjrzyjmy się im bliżej.
Zakres wytycznych
Wytyczne MDCG obejmują zarówno oprogramowanie wyrobów medycznych, jak i oprogramowanie wyrobów medycznych do diagnostyki in vitro (IVD). Zgodnie z dokumentem oprogramowanie wyrobów medycznych (MDSW) definiuje się jako oprogramowanie przeznaczone do stosowania samodzielnie lub w połączeniu z innymi produktami w celu określonym w definicji „wyrobu medycznego” zawartej w wyroby medyczne 2017/745 wyroby medyczne (MDR) lub wyroby medyczne do diagnostyki in vitro 2017/746 (IVDR). Wytyczne określają kryteria, które należy stosować w celu ustalenia, czy oprogramowanie podlegające przeglądowi jest wyrobem medycznym, czy nie, oraz mają na celu dostarczenie dodatkowych wyjaśnień i zaleceń dotyczących MDSW dla producentów wyrobów medycznych i innych stron.
Po pierwsze, wytyczne określają najważniejsze terminy używane w kontekście MDSW, które obejmują:
Zamierzone zastosowanie: zastosowanie, do którego wyrób jest przeznaczony zgodnie z danymi dostarczonymi przez producenta na etykiecie, w instrukcjach użytkowania lub materiałach promocyjnych lub sprzedażowych lub oświadczeniach oraz zgodnie z określeniem producenta w ocenie klinicznej.
Akcesorium: Artykuł, który, choć sam w sobie nie jest wyrobem medycznym, jest przeznaczony przez jego wytwórcę do stosowania wraz z jednym lub kilkoma wyrobami medycznymi, aby w szczególności umożliwić stosowanie wyrobu(ów) medycznego(ych) zgodnie z jego(ich) przewidzianym(i) zastosowaniem(ami) lub aby wspomóc funkcjonalność wyrobu(ów) medycznego(ych) konkretnie i bezpośrednio pod względem jego(ich) przewidzianego(ych) zastosowania(ń). Ponadto MDCG wspomina, że oprogramowanie dodatkowe może napędzać lub wpływać na korzystanie z wyrobu medycznego, a instrukcje użytkowania i inna dokumentacja dostarczona przez producenta powinny zawierać szczegółowe informacje na temat sposobu wyboru odpowiedniego oprogramowania i akcesoriów.
Oprogramowanie: oznacza zestaw instrukcji, które przetwarzają dane wejściowe i tworzą dane wyjściowe.
Określenie oprogramowania urządzenia medycznego
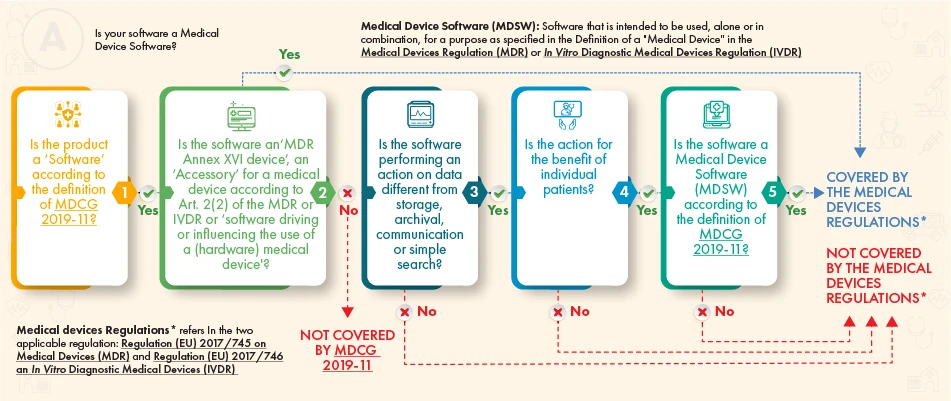
Zgodnie z powyższymi wytycznymi, dane oprogramowanie powinno podlegać regulacji, jeśli spełnia następujące kryteria:
- definicja wyrobu medycznego, jego wyposażenia dodatkowego lub sterowania działaniem wyrobu medycznego, lub
- Wykonuje dodatkowe przetwarzanie danych (nie tylko przechowywanie lub komunikację), a jego działanie przynosi korzyści pacjentom i spełnia definicję oprogramowania wyrobu medycznego zgodnie z wytycznymi MDCG.
wyroby medyczne do diagnostyki in vitro wyroby medyczne do określania
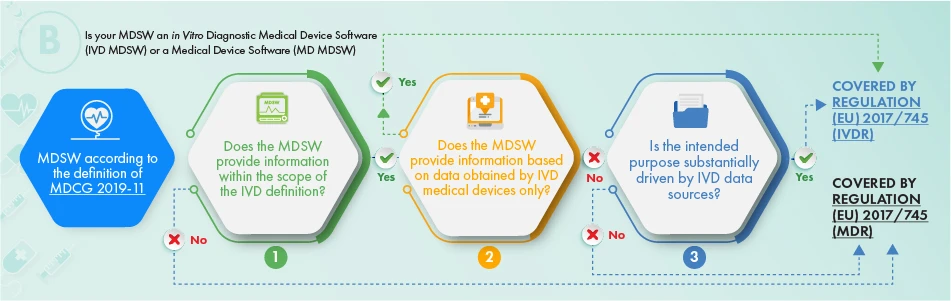
Powyższy schemat opisuje podejście, które należy zastosować w odniesieniu do produktów przeznaczonych do diagnostyki in vitro. Aby ustalić, czy dane oprogramowanie powinno podlegać regulacji, należy uwzględnić następujące kryteria:
- definicja wyrobu medycznego, jego wyposażenia dodatkowego lub sterowania działaniem wyrobu medycznego, lub
- Dostarcza informacji zwykle dostarczanych przez wyroby medyczne do diagnostyki in vitro wyroby medyczne wyłącznie informacji zebranych z wyrobu medycznego do diagnostyki in vitro lub
- Zamierzony cel oprogramowania jest związany z kwestiami IVDR
Zgodnie z wytycznymi MDCG, rodzaj połączenia między oprogramowaniem wyrobu medycznego a urządzeniem nie ma wpływu na kwalifikację oprogramowania jako wyrobu zgodnie z MDR i IVDR. Oprogramowanie wyrobu medycznego może istnieć jako samodzielny produkt lub być wbudowane w urządzenie sprzętowe i wyjaśnia następujące wymogi regulacyjne:
- Biorąc pod uwagę jego kwalifikację i klasyfikację, samodzielne oprogramowanie wyrobu medycznego musi podlegać pełnemu zakresowi procedur regulacyjnych zgodnie z obowiązującymi przepisami.
- Oprogramowanie wyrobu medycznego, które jest integralnym komponentem lub częścią sprzętowego wyrobu medycznego, mogłoby być wprowadzane do obrotu w ramach procedury uproszczonej. Podlegałoby ono przeglądowi nie oddzielnie, ale podczas ogólnej oceny samego sprzętowego wyrobu medycznego.
Podsumowując, wytyczne MDCG obejmują istotne aspekty związane z klasyfikacją oprogramowania wyrobów medycznych oraz określeniem wymogów regulacyjnych, które należy stosować. Producenci wyrobów medycznych, twórcy oprogramowania i inne podmioty muszą przestrzegać i wdrażać zalecenia MDCG, aby zapewnić zgodność z przepisami. Aby uzyskać więcej informacji na temat klasyfikacji software as a medical device, należy skonsultować się z ekspertem ds. regulacji. Bądź na bieżąco. Zachowaj zgodność z przepisami.









