3 min read
Etykietowanie stanowi integralną część marketingu wyroby medyczne. Etykieta to informacja umieszczona na urządzeniu i/lub opakowaniu w formacie czytelnym dla człowieka. Głównym celem etykietowania jest dostarczenie informacji dotyczących bezpieczeństwa użytkownikom, którymi mogą być pracownicy służby zdrowia, konsumenci lub inne odpowiednie osoby.
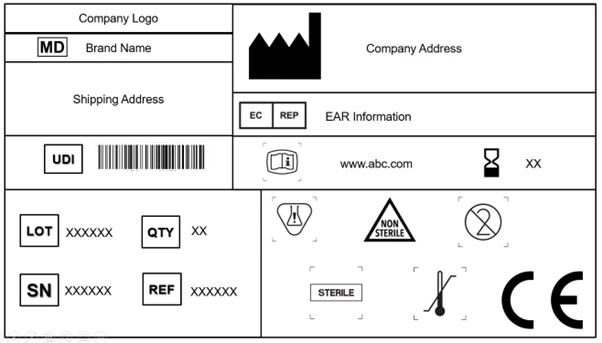
Wszystkie światowe organy regulacyjne mają określone wymagania dotyczące etykietowania. Podobnie UE szczegółowo opisała wymagania dotyczące etykietowania w rozdziale III załącznika I do wyroby medyczne UE wyroby medyczne (EU MDR) 2017/745. Najważniejszą rzeczą, o której należy pamiętać, jest umieszczenie wszystkich symboli zawierających wymagane informacje na etykiecie urządzenia oraz w dokumentach (broszurach, instrukcjach obsługi, instrukcjach użytkowania itp.) dołączonych do niego.
Niektóre z kluczowych kwestii dotyczących etykietowania, na które należy zwrócić uwagę podczas dostosowywania się do wymogów rozporząd EU MDR , to:
1. Symbologia etykietowania urządzeń medycznych
Każdy producent jest zobowiązany do umieszczenia symbolu wyrobu medycznego, który oznacza, że produkt dostarczany na rynek UE jest wyrobem medycznym. Umieszczenie tego symbolu na urządzeniu i wszystkich poziomach opakowania jest obowiązkowe. Ponadto etykieta powinna zawierać nazwę handlową i oryginalną nazwę urządzenia.
2. Urządzenia specjalne
W przypadku, gdy produkt jest urządzeniem specjalnym lub niestandardowym, na etykiecie należy podać jego status. Na przykład, jeśli produkt jest przeznaczony wyłącznie do badań klinicznych, etykieta powinna wyraźnie o tym informować.
W przypadku urządzeń z materiałami chłonnymi lub które mogą lokalnie rozpraszać się w ludzkim ciele, etykieta powinna zawierać skład materiału i szczegóły ilościowe dotyczące kluczowych składników.
W przypadku wyrobów jednorazowego użytku i wyrobów sterylnych wymagane jest nawet wyraźne oznakowanie. W przypadku wyrobów poddanych ponownemu przetworzeniu etykieta powinna zawierać informację o tym, ile razy można poddać je ponownemu przetworzeniu, ile razy zostały one poddane ponownemu przetworzeniu do tej pory oraz o zastosowanej metodzie sterylizacji.
3. Obecność substancji toksycznych
Deklaracja obecności substancji CMR (rakotwórczych, mutagennych, działających szkodliwie na rozrodczość) i substancji zaburzających gospodarkę hormonalną jest obowiązkowa na etykietach, jeśli stężenie przekracza 0,1% w/w. Lista takich substancji musi być dołączona do wyrobu i/lub opakowania.
Ponadto na wyrobach musi być umieszczona etykieta informująca o obecności pochodnych krwi i tkanek (nawet jeśli są one zawarte w substancji leczniczej wyrobu złożonego).
4. Zharmonizowane normy
Rozporządzenie EU MDR uznaje i akceptuje normę ISO 15223-1: 2021. Dokument określa symbole, które należy stosować w oznakowaniu wyroby medyczne ich opakowań. Rozdział 3 (23.1,h) załącznika I do rozporządzenia EU MDR że można stosować symbole uznane na arenie międzynarodowej, a w przypadku regionów, w których symbole te nie są uznawane, wymagane jest dostarczenie opisu tych symboli w dokumencie dołączonym do wyrobu.
5. UDI
Artykuły 27, 28, 29 i załącznik VI (A, B, C) szczegółowo określają zasady i przepisy dotyczące kodu UDI. Etykieta musi teraz zawierać nośnik kodu UDI [automatyczna identyfikacja do przechwytywania danych (AIDC) i czytelna dla człowieka reprezentacja kodu UDI (HRI)] na urządzeniu i wyższych poziomach opakowania. Wyższe opakowanie urządzenia (z wyłączeniem opakowań wysyłkowych) będzie miało własny nośnik kodu UDI.
6. Elektroniczne informacje do użytku (eIFU)
Adres internetowy (URL) w formie eIFU może być również umieszczony na etykiecie wyrobu medycznego wraz z papierową instrukcją użycia. eIFU może być stosowana w przypadku wyroby medyczne wszczepialnych, aktywnych wyroby medyczne wszczepialnych, wyroby medyczne stałych wyroby medyczne oprogramowania (przeznaczonego również dla osób nieposiadających wiedzy specjalistycznej).
7. Informacje o wykonawcach (EO)
Etykieta zwykle zawiera informacje o producencie. Jednakże, w przypadku zagranicznych producentów, informacje autoryzowanego przedstawiciela powinny być umieszczone na etykietach handlowych.
8. Ostrzeżenia i środki ostrożności
Ostrzeżenia i środki ostrożności muszą być wymienione na etykiecie urządzenia. Informacje dotyczące tego aspektu mogą być ograniczone do minimum, a ich szczegóły można podać w instrukcji obsługi.
Producenci są również zobowiązani do dostosowania się do krajowych wymogów dotyczących etykietowania. Wymogi językowe zależą od państwa członkowskiego UE. Może to mieć duży wpływ na etykietowanie urządzenia, IFU i opakowanie pod względem czasu i kosztów.
Te dodatkowe wymagania mogą dodatkowo zwiększyć obciążenie producenta w związku z istniejącą złożonością procesu etykietowania. Niepowodzenie w tym zakresie może stać się bardzo kosztowne, obejmując wycofanie produktu z rynku i kolejne etapy działań naprawczych i zapobiegawczych (CAPA).
Szukasz pomocy w zakresie etykietowania zgodnie z rozporządzeniem EU MDR? Freyr kompleksowe usługi w zakresie etykietowania wyroby medyczne. Skontaktuj się z naszymi ekspertami ds. regulacyjnych już teraz pod adresem – sales@freyrsolutions.com









