
3 min read
510(k) to przed wprowadzeniem na rynek wniosek składany do FDA wykazania, że urządzenie, które ma być wprowadzone do obrotu, jest tak samo bezpieczne i skuteczne, to znaczy zasadniczo równoważne z urządzeniem legalnie wprowadzonym do obrotu (urządzeniem referencyjnym). Urządzenia o umiarkowanym ryzyku wymagają złożenia powiadomienia 510(k), które obejmuje niewielką część urządzeń klasy I i III oraz większość urządzeń klasy II.
Istnieją trzy (03) rodzaje programów 510(k): tradycyjny, skrócony i specjalny. Ścieżka bezpieczeństwa i wydajności została wprowadzona w 2019 r. i opiera się na programie skróconym. Program eSTAR wprowadzony w 2020 r. umożliwia kompleksowe zgłoszenie wyrobu medycznego za pośrednictwem interaktywnego formularza PDF.
Kto potrzebuje certyfikatu 510(k)?
510(k) to zasadniczo nazwa procesu/ścieżki, którą US producenci urządzeń medycznych zamierzający wprowadzić na rynek US swoje urządzenia o średnim lub wysokim ryzyku, aby wykazać, że produkt, który ma być wprowadzony do obrotu, jest tak samo bezpieczny i skuteczny jak urządzenie wprowadzone do obrotu zgodnie z prawem.
Poniżej opisano krok po kroku proces uzyskiwania zezwolenia 510(k).
Krok 1- Identyfikacja kodu klasy urządzenia, typu zgłoszenia i urządzenia źródłowego
- Określ kod produktu i numer regulacji — aby określić wymagania testowe 510(k), należy najpierw określić kod produktu i numer regulacji. Można rozpocząć wyszukiwanie w FDA , aby znaleźć 7-cyfrowy numer regulacji, którego identyfikacja odpowiada przeznaczeniu danego urządzenia.
- Kod FDA składa się z trzech (03) liter. Za pomocą tego kodu można znaleźć informacje dotyczące klasyfikacji produktu, opisu przepisów i wymagań GMP.
- Wybór rodzaju wniosku – wnioskodawca może wybrać jeden z trzech (03) rodzajów wniosków wymienionych powyżej. Tradycyjny 510(k) jest przeznaczony dla osób składających wniosek po raz pierwszy, specjalny 510(k) jest przeznaczony dla producentów urządzeń medycznych, którzy chcą zgłosić zmiany w istniejącym urządzeniu, a skrócony 510(k) można wybrać, gdy urządzenie jest zgodne z ustalonymi dobrowolnymi normami konsensusowymi. W przypadku skróconego 510(k) osoba składająca wniosek musi opierać się na FDA .
- Identyfikacja urządzenia źródłowego - producent urządzenia medycznego musi udowodnić, że urządzenie, które zamierza wprowadzić do obrotu, ma takie samo zamierzone zastosowanie i właściwości techniczne jak legalnie wprowadzone do obrotu urządzenie, znane również jako urządzenie źródłowe. Jeśli istnieją jakiekolwiek różnice w charakterystyce technicznej, zgłaszający musi udowodnić, że z różnicą tą nie wiążą się żadne obawy dotyczące bezpieczeństwa i skuteczności.
Krok 2- Przygotowanie pliku 510(k)
Kolejnym krokiem jest przygotowanie dokumentacji 510(k), wytycznych i informacji, które są dostępne na FDA . Zawiera ona listę kontrolną akceptacji dla wszystkich trzech (03) rodzajów programów 510(k) oraz mikrostronę zatytułowaną „Treść dla 510(k)”, która zawiera informacje dotyczące oświadczeń dotyczących wskazań do stosowania, porównania istotnej równoważności i proponowanego oznakowania, a także inne przydatne informacje.
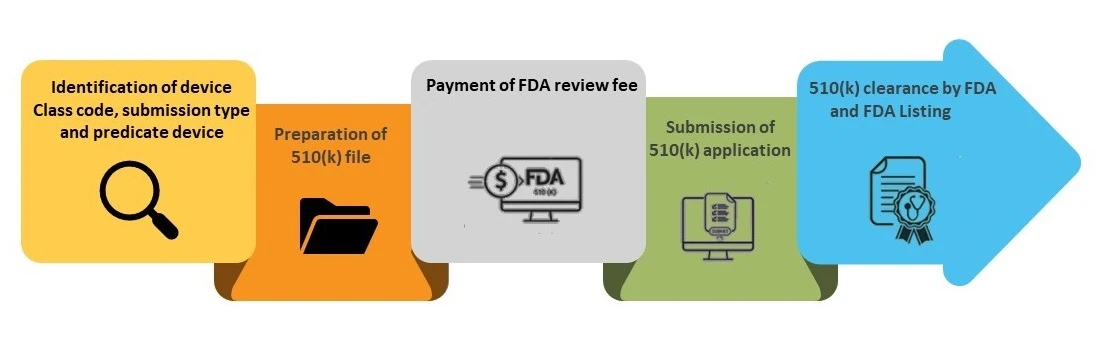
Etapy procesu składania wniosku 510(k)
Krok 3 – Uiszczenie opłaty za FDA
Wszystkie rodzaje wniosków 510(k) podlegają opłacie użytkownika. W roku finansowym 2023 standardowa opłata za 510(k) wynosi 19 870 USD. W przypadku firm certyfikowanych przez Centrum Diagnostyki i Zdrowia Radiologicznego (CDRH), znanych również jako małe firmy, opłata wynosi 4 967 USD. Opłata może ulec zmianie w następnym roku finansowym.
Krok 4 - Złożenie wniosku 510(k)
Podmiot składający wniosek może wysłać elektroniczną kopię (eCopy) lub elektroniczny szablon wniosku i zasobów (eSTAR) za pośrednictwem portalu CDRH.
Począwszy od 01 października 2023 r. wszystkie zgłoszenia 510(k), o ile nie są wyłączone zgodnie z ostatecznymi wytycznymi, muszą być składane w formie elektronicznej za pomocą eSTAR.
Po złożeniu wniosku 510(k) przypisywany jest unikalny numer kontrolny, znany jako „numer 510(k)” lub „numer K”. FDA dwie kontrole weryfikacyjne: jedną w celu sprawdzenia, czy uiszczono odpowiednią opłatę użytkownika, a drugą w celu sprawdzenia, czy dostarczono ważną kopię elektroniczną lub eSTAR.
- W ciągu 7 dni FDA potwierdzenie odbioru, jeśli uiszczono odpowiednią opłatę użytkownika i dostarczono ważną kopię elektroniczną lub eSTAR. W przeciwnym razie FDA pismo wstrzymujące dotyczące nierozwiązanych kwestii.
- W ciągu 15 dni FDA ocenę akceptacyjną . FDA wnioskodawcę , czy wniosek 510(k) został przyjęty do oceny merytorycznej, czy też został odrzucony (RTA).
- W ciągu 60 dni FDA merytoryczną ocenę. FDA za pośrednictwem interaktywnej wymiany informacji (
) o FDA interaktywnej oceny lub wstrzymaniu procedury 510(k) i zażądaniu dodatkowych informacji.
Krok 5 – FDA i wpisanie do bazy danych FDA (k)
Celem FDA ogłoszenie decyzji w sprawie poprawek do opłat za użytkowanie wyrobów medycznych (MDUFA) w ciągu 90 FDA . FDA to dni kalendarzowe między datą otrzymania wniosku 510(k) a datą podjęcia decyzji MDUFA, z wyłączeniem dni, w których wniosek był wstrzymany z powodu prośby o dodatkowe informacje. Decyzje MDUFA dotyczące wniosków 510(k) obejmują ustalenia dotyczące istotnej równoważności (SE) lub braku istotnej równoważności (NSE).
Po podjęciu decyzji FDA do wnioskodawcy pismo z decyzją pocztą elektroniczną. Wniosek 510(k), który otrzymał pismo z decyzją SE, uznaje się za „zatwierdzony”. Następnie jest on umieszczany w bazie danych 510(k) wraz z wskazaniami do stosowania wyrobu medycznego oraz podsumowaniem 510(k) lub oświadczeniem 510(k) w formie załączników.
Można stwierdzić, że staranne planowanie i realizacja poprzez dokładną dokumentację oraz dogłębne zrozumienie otoczenia regulacyjnego mają kluczowe znaczenie dla pomyślnego złożenia wniosku 510(k) do FDA.
Aby uzyskać pomoc w procesie składania wniosku 510(k) dotyczącego urządzenia medycznego, można napisać do us adres sales@freyrsoltions.com lub umówić się na rozmowę telefoniczną z naszymi ekspertami, którzy pomogą Państwu przejść przez procedury. Bądź na bieżąco. Zachowaj zgodność z przepisami.









