
2 min read
wyroby medyczne przed umieszczeniem na liście ARTG wyroby medyczne ocenie zgodności w celu zapewnienia, że są one zgodne z podstawowymi zasadami wymaganymi przez australijską agencję Therapeutic Goods Administration (TGA). Podstawowe zasady określają zasadniczo cechy bezpieczeństwa i działania, które powinien spełniać każdy wyrób, aby mógł być sprzedawany w Australii. Podobnie jak w przypadku procesu obowiązującego w UE, ocena zgodności w Australii opiera się na klasie ryzyka danego wyrobu. W związku z tym ścieżka oceny zgodności, którą należy zastosować w przypadku każdego urządzenia, zależy od jego klasyfikacji.
Istnieją różne normy regulacyjne, które mają zastosowanie do różnych typów urządzeń. Spełnienie jednej lub kilku norm jest wymagane jako podstawowy wymóg. Niezbędnym warunkiem wstępnym jest identyfikacja odpowiednich norm mających zastosowanie do urządzenia, a następnie przetestowanie urządzenia w celu wykazania jego zgodności z normą.
Ocena zgodności obejmuje systematyczne badanie dokumentacji technicznej dotyczącej wyrobu. Kluczowymi obszarami oceny są zarządzanie ryzykiem, ocena kliniczna, proces produkcji oraz działania w zakresie nadzoru prowadzone przez producenta. Wyroby, które muszą posiadać certyfikat zgodności TGA, są wymienione w przepisie 4.1 rozporządzenia w sprawiewyroby medyczne(Therapeutic Goods (wyroby medyczne) Regulations) z 2002 r. Chociaż certyfikat oceny zgodności wydany przez TGA jest warunkiem wstępnym wprowadzenia do obrotu większości wyroby medyczne Australii, TGA akceptuje również certyfikaty oceny zgodności wydane przez jednostki notyfikowane w Europie. Ponadto TGA akceptuje również oceny zgodności z krajów należących do MDSAP Medical Device Single Audit Program).
Wymagania dotyczące oceny zgodności różnią się w zależności od kategorii ryzyka urządzenia. Tabela nr 1 zawiera szczegółowe informacje na temat procesu oceny zgodności opartego na klasyfikacji w Australii.
Proces oceny zgodności – Australia
Klasa | Procedura oceny zgodności | Obowiązki producenta |
| I | Część 6 (Deklaracja zgodności, niewymagająca oceny przez sekretarza) | Dokumentacja potwierdzająca zgodność z zasadami podstawowymi |
| I (mierzący) i IIa (niesterylny) | Część 6 (Deklaracja zgodności, niewymagająca oceny przez sekretarza) Część 5 (System zarządzania jakością produktu) | Dokumentacja potwierdzająca zgodność z zasadami podstawowymi. Wdrożenie systemu zarządzania jakością produktów w zakresie kontroli końcowej i testowania na potrzeby audytów. Uwaga: Przedłożenie deklaracji zgodności nie jest wymagane dla klasy I (produkty niemierzące i niesterylne), ale powinna być ona dostępna na żądanie TGA. |
| I (sterylny) i IIa (sterylny) | Część 6 (Deklaracja zgodności, niewymagająca oceny przez sekretarza) Część 4 (Zapewnienie jakości produkcji) | Dokumentacja potwierdzająca zgodność z zasadami podstawowymi. Wdrożenie systemu zarządzania jakością, który wyklucza elementy projektowe, w oparciu o ISO 13485. |
| IIb | Część 1 (Pełna kontrola jakości) z wyłączeniem punktu 1.6 (Badanie projektu) | Wdrożenie pełnego systemu zarządzania jakością obejmującego projektowanie, produkcję, etykietowanie, pakowanie i kontrolę końcową w oparciu o ISO 13485. Nie obejmuje dokumentacji projektowej. |
| III i AIMD | Część 1 (Pełna kontrola jakości) wraz z punktem 1.6 (Badanie projektu) | Wdrożenie pełnego systemu zarządzania jakością obejmującego projektowanie, produkcję, etykietowanie, pakowanie i kontrolę końcową w oparciu o ISO 13485. Dokumentacja projektowa zgodna z podstawowymi zasadami. |
| Systemy lub pakiety procedur Część 7 | (Procedury dotyczące wyroby medyczne do specjalnych zastosowań) | Podstawowe zasady. Procedura oceny zgodności. Dowody kliniczne dotyczące poszczególnych składników systemu lub opakowania. |
Tabela nr 1. Wymagania dotyczące oceny zgodności urządzeń
Zwykły proces zatwierdzania wyroby medyczne TGA wygląda następująco:
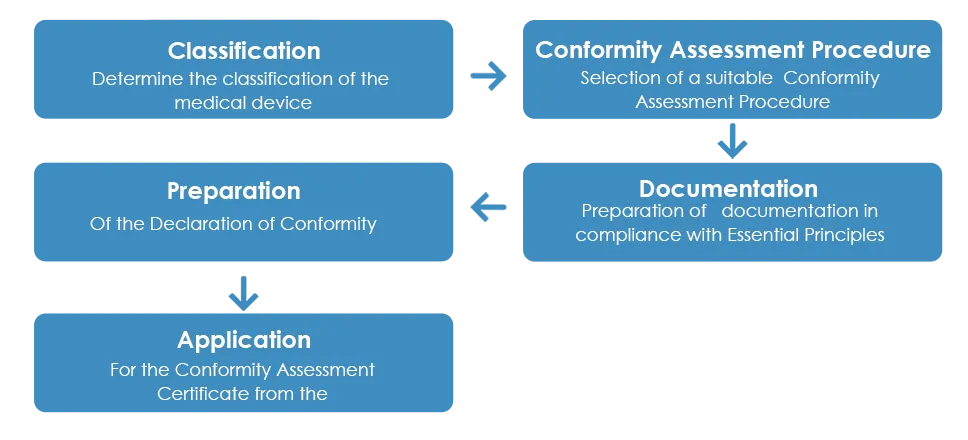
Jak opisano powyżej, po otrzymaniu certyfikatu oceny zgodności od TGA producent musi przygotować deklarację zgodności (DoC), w której stwierdza, że wyrób medyczny jest zgodny z obowiązującymi zasadami zasadniczymi, zasadami klasyfikacji i ścieżką oceny zgodności. Jednak w przeciwieństwie do certyfikatu oceny zgodności, europejska deklaracja zgodności nie jest akceptowana przez TGA.
Rynek australijski oferuje obiecujące perspektywy dla producentów urządzeń medycznych, o ile spełnione są wymogi regulacyjne Therapeutic Goods Administration (TGA). Wymogi dotyczące zgodności i proces oceny różnią się w zależności od klasy ryzyka urządzenia i IVD. Chociaż przepisy dotyczące urządzeń medycznych są dobrze zdefiniowane i przejrzyste, poruszanie się po nich jest skomplikowane, a producenci mogą zwrócić się o pomoc do partnerów regulacyjnych, aby pomyślnie wprowadzić urządzenie na rynek.
Aby uzyskać wyczerpujące informacje na temat wymagań dotyczących oceny zgodności, australijskiego sponsora i rejestracji w ARTG w Australii,skontaktuj się z doświadczonym ekspertem ds. regulacji. Bądź na bieżąco. Dbaj o zgodność z przepisami.









