
2 min de lectura
Tras el descubrimiento inicial de impurezas de nitrosamina en medicamentos y principios activos farmacéuticos (APIs) por parte de USFDA mediados de 2018, los organismos reguladores de la UE se han sumado a muchos otros países en un intento por prevenir los riesgos que conlleva. Han retirado del mercado varios medicamentos que suponen un peligro para la salud debido a esta sustancia. Conocida por sus propiedades cancerígenas, la nitrosamina puede resultar perjudicial cuando se ingiere por encima de los niveles aceptados para los seres humanos. La sustancia N-nitrosodimetilamina (NDMA) tiene un límite aceptable de 96 ng/día y cualquier cantidad superior a esta es inaceptable en medicamentos e APIs.
Las nitrosaminas se forman cuando las aminas secundarias, terciarias o cuaternarias reaccionan con un agente nitrosante. Se están comprobando las impurezas de todos los medicamentos que contienen principios activos sintetizados químicamente. El Comité de Medicamentos de Uso Humano (CHMP) ha revisado y elaborado un informe de evaluación. Ha solicitado a los titulares de autorizaciones de comercialización (MAH) que sigan las últimas directrices para revisar todos los medicamentos químicos y biológicos actualmente disponibles en el mercado para consumo humano.
Los titulares de autorizaciones de comercialización son responsables de garantizar que el proceso de fabricación de todos los productos biológicos y químicos se revise periódicamente para identificar cualquier contaminación. Una vez identificada, se deben tomar las medidas pertinentes para mitigar los riesgos que plantea. Aunque las posibilidades de que se produzca dicha contaminación son menores durante la fabricación de medicamentos químicos y biológicos, la Agencia Europea de Medicamentos (EMA) no se arriesga. Las empresas farmacéuticas deben garantizar que se aplican los protocolos de fabricación pertinentes de acuerdo con las últimas EMA . También son responsables de comprobar los niveles de nitrosamina en los medicamentos disponibles en el mercado y de mantenerlos dentro de los límites aceptables.
Tras finalizar una revisión en virtud del artículo 5, apartado 3, del Reglamento (CE) n.º 726/2004 (solicitud de revisión) el año pasado, la EMA publicado nuevas directrices para evitar la presencia de contaminación por nitrosaminas en los medicamentos de uso humano. El proceso es el mismo para los productos autorizados a nivel nacional y los autorizados a nivel central. La EMA, junto con la Dirección Europea de Calidad del Medicamento y de la Atención Sanitaria (EDQM), aplicará el artículo 5, apartado 3, del CHMP. A continuación se describe el proceso que debe seguirse para cumplir con las modificaciones.
La guía en tres etapas para los HMA
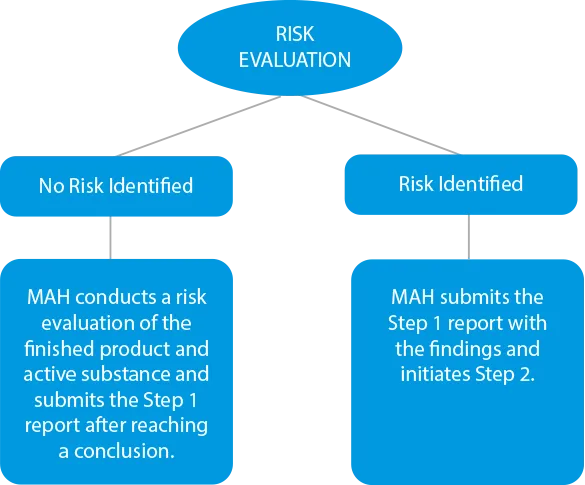
- Evaluación de riesgos - Los fabricantes deben llevar a cabo un proceso de evaluación de riesgos para identificar las sustancias activas y el producto acabado para comprobar los niveles de nitrosamina. Si hay algún caso de contaminación cruzada, también debe incluirse en el informe de resultados. Los plazos de presentación en esta fase se fijaron el 31 de marzo de 2021 para los medicamentos químicos y el 1 de julio de 2021 para los biológicos.
![]()
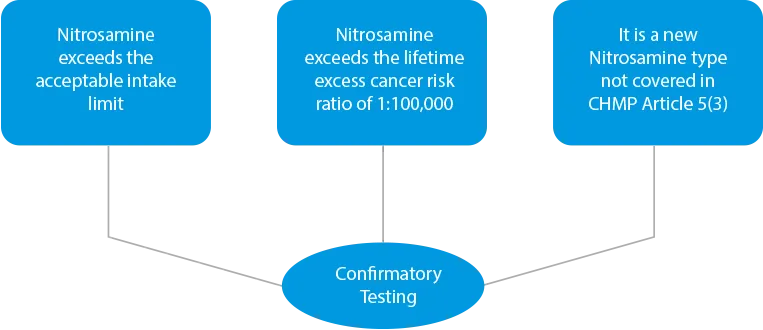
- Pruebas de confirmación - En caso de detectarse contaminación cruzada o si los productos se identifican como peligrosos debido a la presencia de niveles más altos de nitrosamina, es necesario realizar pruebas de confirmación. Las pruebas de confirmación son obligatorias en tres (03) casos.
![]()
- Modificaciones de la autorización de comercialización: cuando se detecta la presencia de nitrosamina, es necesario realizar dos (02) pruebas confirmatorias para comunicar los resultados correctos a la EMA. Sobre la base de estos resultados, los titulares de la autorización de comercialización deben solicitar un cambio en el proceso de fabricación. Esto se hace mediante los procedimientos reglamentarios estándar, con la ayuda de una variación de la plantilla de autorización de comercialización. Los plazos para ello son el 26 de septiembre de 2022 para los productos químicos y el 1 de julio de 2023 para los medicamentos biológicos.
El camino ideal para que los HMA afronten el cumplimiento del nuevo nivel de nitrosaminas
Dado que se trata de la última novedad EMA la EMA para mitigar los riesgos relacionados con los niveles de nitrosamina en los medicamentos y las situaciones de contaminación cruzada, todo el proceso puede resultar bastante abrumador para los titulares de autorizaciones de comercialización y los fabricantes de principios activos farmacéuticos. Ya sea para presentar la plantilla de respuesta cuando se detecta contaminación en el paso 1 o para realizar las pruebas correspondientes, todas las fases deben cumplir con las nuevas normas. Los titulares de autorizaciones de comercialización deben colaborar con expertos en regulación que estén al día de los últimos cambios y garantizar el cumplimiento de las nuevas directrices. Elija al socio adecuado, como Freyr evitar retrasos y errores.









