
5 min de lectura
Dispositivos Médicos en Corea del Sur se utiliza para diagnosticar, tratar y supervisar a los pacientes en el sistema sanitario moderno. Comprende tanto software integrado en dispositivos médicos como software independiente que puede utilizarse en ordenadores personales, dispositivos móviles y servicios basados en la web. El Ministerio de Seguridad Alimentaria y Farmacéutica (MFDS) de Corea del Sur es responsable de regular Dispositivos Médicos y garantizar su seguridad y eficacia. El 5 de julio de 2023, el MFDS estableció los criterios para la aprobación e inspección del Dispositivos Médicos ; estas normas proporcionan una estructura que los solicitantes civiles pueden seguir al presentar el software para su aprobación o revisión.
Las regulaciones abordan una variedad de temas, incluyendo el alcance de la aplicación, los requisitos de documentación técnica y los informes de verificación del cumplimiento. Existen normas y directrices internacionales que se aplican al Dispositivos Médicos , además de las directrices de la MFDS, como la norma 62304 de la Comisión Electrotécnica Internacional (IEC) para los procesos del ciclo de vida del software y la guía de la Administración de Alimentos y Medicamentos de los Estados Unidos (US FDA) sobre aplicaciones médicas móviles.
Plan de desarrollo de software y análisis de requisitos
- El Plan de Desarrollo de Software describe el enfoque general para el desarrollo de software, incluyendo especificaciones, métodos y herramientas de desarrollo. También abarca la verificación, la gestión Dispositivos Médicos , la gestión de la configuración y la documentación.
- El análisis de requisitos establece los requisitos Dispositivos Médicos , incluidas las medidas de control de riesgos y los métodos de verificación. Mediante una planificación y un análisis minuciosos del proceso de desarrollo del software, los desarrolladores pueden garantizar que el software resultante cumpla con los estándares necesarios de seguridad y eficacia.
- El informe de verificación de conformidad del software incluye un resumen del plan de desarrollo del software, el número de control de documentos del fabricante y una descripción general del análisis de requisitos. Al cumplir estas directrices, Dispositivos Médicos se puede desarrollar con confianza, sabiendo que ha sido sometido a rigurosas pruebas y que cumple con los estándares necesarios de seguridad y eficacia.
Dispositivos Médicos Verificación y validación de Dispositivos Médicos
- La verificaciónDispositivos Médicos garantiza que el software cumpla con los requisitos especificados.
- La validaciónDispositivos Médicos garantiza que el software satisfaga las necesidades del usuario y los usos previstos.
- El informe de verificación y validaciónDispositivos Médicos describe el proceso de verificación y validación, incluyendo el nombre del producto, la revisión y los nombres de las personas que examinaron y aprobaron el informe. El informe puede variar en función de las características del software, pero debe incluir una descripción del software, los métodos de verificación y validación utilizados y los resultados de las pruebas.
Entorno operativo y software de procedencia desconocida (SOUP)
- Si el software depende de un hardware específico, como el software integrado, el documento técnico debe describir las especificaciones del hardware.
- Sin embargo, si el software es independiente y está desarrollado para funcionar en hardware de uso general, el entorno operativo debe describirse en la materia prima. Esto incluye las especificaciones mínimas recomendadas, como Microsoft Windows 10 o superior.
- Además, si el Dispositivos Médicos incluye software comercial de procedencia desconocida (SOUP), se debe crear un entorno operativo que garantice su correcto funcionamiento. Al describir cuidadosamente el entorno operativo y abordar cualquier SOUP, los desarrolladores pueden garantizar que su Dispositivos Médicos sea seguro y eficaz para el uso previsto.
Requisitos de gestión Dispositivos Médicos y documentación de Dispositivos Médicos
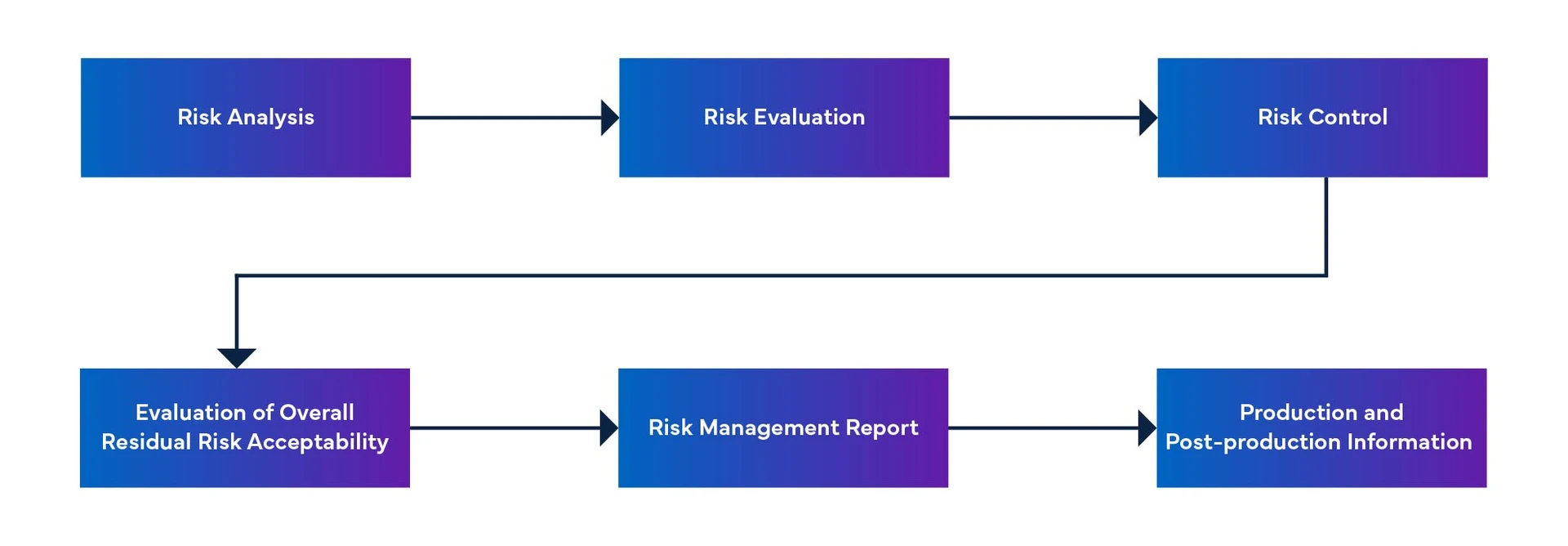
- El proceso de gestión Dispositivos Médicos del software como Dispositivos Médicos incluye la identificación de situaciones peligrosas, el establecimiento de medidas de control de riesgos, la verificación de dichas medidas y la gestión de los cambios en el software.
- El documento de gestión de riesgos de software MFDS-RM proporciona información sobre la gestión de riesgos de software.
- Además, los requisitos de documentación son esenciales para garantizar que el software cumple las normas necesarias de seguridad y eficacia.
- El plan de desarrollo del software, el análisis de requisitos Dispositivos Médicos y los informes de verificación y validación del software deben incluirse en la documentación.
- El Informe de Verificación de la Conformidad del Software describe los requisitos de documentación; también incluye un resumen de los documentos aplicables y el número de control de documentos del fabricante.
Figura 1: Proceso de gestión Dispositivos Médicos
Anomalías no resueltas y medidas correctoras para el software SaMD
- El documento MFDS-PR (Resolución de problemas de software) describe el proceso de resolución de problemas de software, que incluye la notificación, el análisis, la aplicación y la verificación de los problemas.
- El documento también incluye una lista de problemas, fallos, defectos y anomalías sin resolver, así como una evaluación del riesgo residual del sistema de software.
- Las medidas correctoras adoptadas para solucionar estos problemas deben documentarse en el plan de mantenimiento del software, que se establece de acuerdo con el proceso de mantenimiento del software.
- El documento de mantenimiento de MFDS proporciona información sobre SaMD y la resolución de problemas.
Requisitos de revisión y presentación de documentos técnicos para el software SaMD
Los principales documentos de revisión durante el proceso de revisión son los datos de rendimiento, el informe de confirmación de conformidad y los datos de verificación y validación Dispositivos Médicos , la Especificación de Diseño de Software (SDS), la Declaración de Requisitos Dispositivos Médicos (SRS) y los informes de verificación y validación. Se deben presentar el Informe de Confirmación de Conformidad y el Informe de Verificación y Validación Dispositivos Médicos .
Dispositivos Médicos Gestión de riesgos Dispositivos Médicos
- Identificar los peligros potenciales asociados al software y a su uso.
- Evaluar la gravedad de los riesgos asociados a estos peligros.
- Aplicar medidas de control de riesgos para minimizar la probabilidad de daños.
- Seguimiento y revisión de la eficacia de estas medidas de control de riesgos.
- Documentar todas las actividades y decisiones relacionadas con la gestión Dispositivos Médicos .
En un sistema de software, los elementos de software se dividen en partes más pequeñas, incluidos los elementos de software detallados. Cuando un elemento no puede desglosarse más, se denomina unidad. El sistema permite desglosar hasta el nivel de unidad, lo que ayuda a determinar el nivel de seguridad de cada elemento de software. Al reunir estos elementos de software, podemos calcular el nivel de seguridad de todo el sistema de software.
Figura 2: Desmontaje e integración Dispositivos Médicos
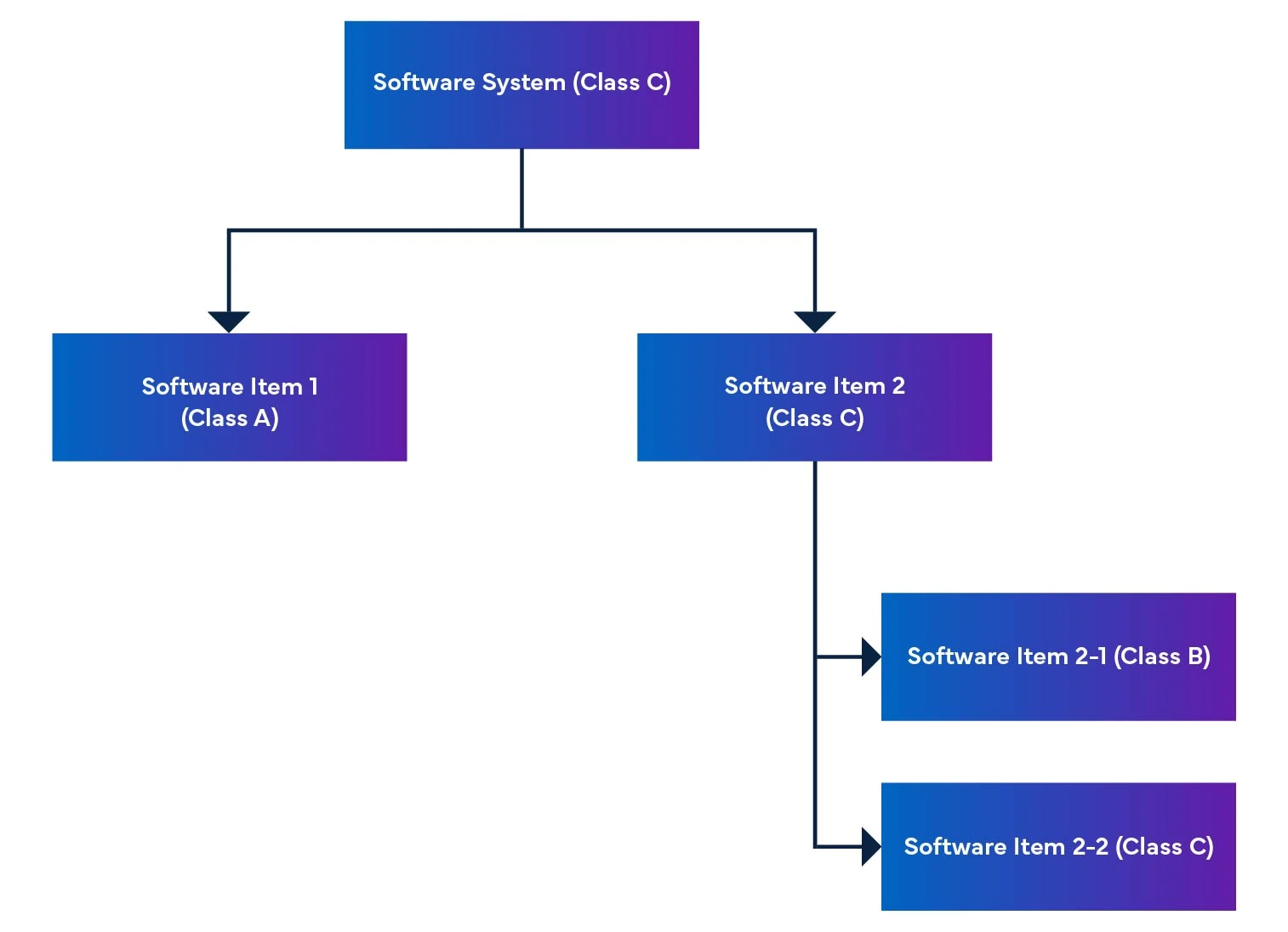
El reglamento también menciona la calificación de seguridad del software, que es una calificación para identificar los riesgos del software SaMD (véase la Tabla 1).
Tabla 1: Definición de la calificación de seguridad
| Clasificación | Dispositivos Médicos Definición de clase de seguridad Dispositivos Médicos |
| Clase A | No hay posibilidad de lesiones o daños corporales. |
| Clase B | Es probable que se produzcan lesiones menos graves (lesiones leves). |
| Clase C | Posibilidad de lesiones graves o muerte. |
Gestión de la configuración del software
- Mantener una documentación precisa y actualizada de todas las versiones, cambios y actualizaciones del software.
- Garantizar que toda la documentación se revisa y aprueba adecuadamente.
- Implantación de procedimientos para gestionar los cambios de configuración del software.
- Documentar todas las actividades y decisiones de gestión de la configuración del software.
Mantenimiento de software
- Probar y supervisar periódicamente el software para garantizar que sigue siendo seguro y eficaz para el uso previsto.
- Aplicar procedimientos para resolver los problemas que puedan surgir, incluidas las correcciones de errores y las actualizaciones de software.
- Documentar todas las actividades y decisiones de mantenimiento del software.
Solución de problemas
- Identificar la causa raíz del problema.
- Aplicar medidas correctoras para resolver el problema.
- Documentar todo el proceso de resolución de problemas para futuras consultas.
Siguiendo las directrices anteriores, los desarrolladores pueden garantizar que cualquier problema relacionado con su Dispositivos Médicos se aborde y documente adecuadamente, y que el software cumpla los requisitos necesarios para su aprobación o examen.
Si usted es un Dispositivos Médicos que desea cumplir con las normas Dispositivos Médicos de Corea del Sur, los expertos en regulación de Freyr pueden guiarle a través del complejo panorama normativo del país. Nos aseguraremos de que sus dispositivos se ajusten a las últimas Dispositivos Médicos de Corea del Sur en materia Dispositivos Médicos para garantizar un cumplimiento perfecto. ¡Póngase en contacto con nosotros para obtener más información!









