
2 min de lectura
FDA US FDA publicado un documento orientativo que ayudará a la industria y al personal de la Agencia de Salud (HA) a determinar cuándo un cambio de software en un Dispositivos Médicos el fabricante presente y obtenga la FDA de una nueva notificación previa a la comercialización (510(k)). Esta guía tiene por objeto mejorar la previsibilidad, la coherencia y la transparencia del proceso de toma de decisiones sobre «cuándo presentar» la notificación, proporcionando un enfoque menos gravoso y describiendo el marco normativo, las políticas y las prácticas que subyacen a dicha decisión, específicamente en relación con los cambios de software. Veamos en detalle la FDA .
Principios rectores y diagrama de flujo FDA
Con la intención de ayudar a Dispositivos Médicos a aplicar los principios fundamentales, el documento proporciona un diagrama de flujo, aclaraciones adicionales y ejemplos necesarios para tomar decisiones sobre una nueva notificación previa a la comercialización 510(k) para un cambio de software realizado en un dispositivo ya aprobado en los US. Además, se deben seguir varios principios rectores al utilizar esta guía para determinar si se debe presentar una nueva 510(k) para cambiar un dispositivo existente. Algunos de ellos son ampliamente conocidos y se derivan de la política actual FDA (k), mientras que otros son necesarios para utilizar el esquema lógico mencionado en esta guía. Según la guía, el esquema proporcionado no puede abarcar todas las posibles complejidades relacionadas con dichos cambios y cómo afectan a la decisión. Por lo tanto, para determinar la necesidad de una nueva notificación previa a la comercialización 510(k), Dispositivos Médicos deben tener en cuenta los principios generales y el diagrama de flujo que se resumen a continuación.
- Cambios que afectan a la seguridad o eficacia de un producto
- Evaluación inicial basada en el riesgo
- Consecuencias imprevistas de los cambios
- Utilización de la gestión de riesgos
- Función de las pruebas (actividades de verificación y validación) en la evaluación de si un cambio podría afectar significativamente a la seguridad y la eficacia.
- Evaluar los cambios simultáneos para determinar si es necesaria la presentación de un nuevo 510(k).
- Dispositivo comparativo adecuado y efecto acumulativo de los cambios
- Requisitos de documentación (21 CFR Parte 820)
- Presentaciones 510(k) de productos modificados
- Determinaciones de equivalencia sustancial
- Es probable que se requiera la presentación de un nuevo 510(k) si un fabricante modifica su dispositivo para afectar a la seguridad o eficacia del mismo. Sin embargo, los cambios que no pretendan afectar a la seguridad o eficacia de un producto deben evaluarse igualmente.
- Para determinar si un cambio o modificación podría afectar significativamente a la seguridad o eficacia, el fabricante debe realizar primero una evaluación basada en el riesgo para saber si el cambio podría afectar positiva o negativamente a la seguridad o eficacia del producto. Esta evaluación basada en el riesgo debe identificar y analizar todos los riesgos nuevos y los cambios en los riesgos existentes derivados del cambio del producto y conducir a una decisión inicial sobre si es necesaria la presentación de un nuevo 510(k).
- A veces hay consecuencias adicionales no previstas o imprevistas que pueden desencadenarse durante la presentación de software. El diagrama de flujo debe evaluar estas consecuencias para determinar si es necesaria la presentación de un nuevo 510(k).
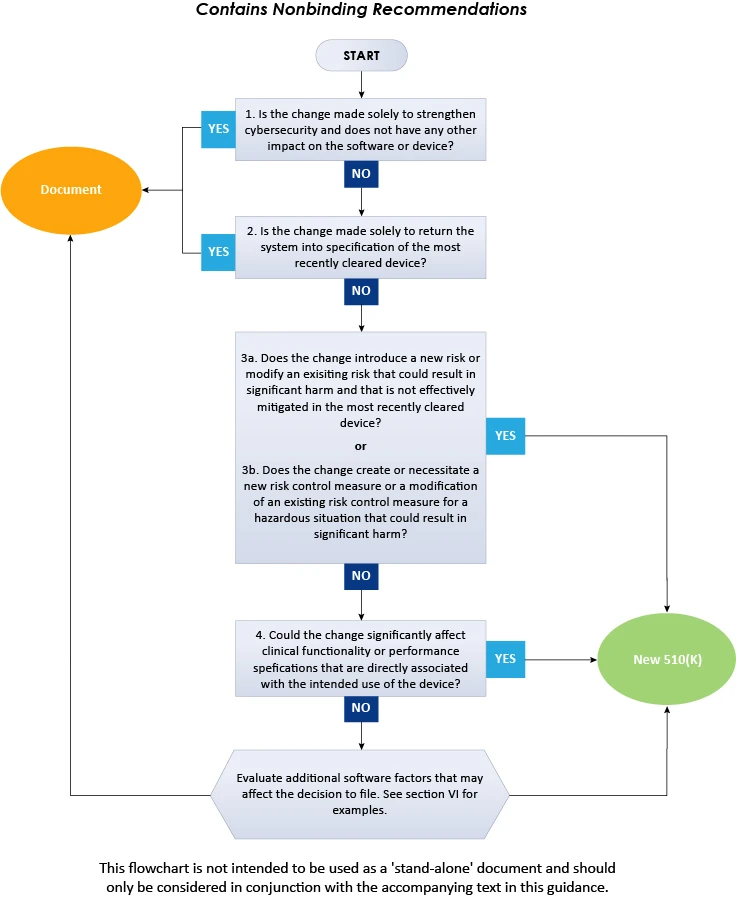
El diagrama de flujo anterior ilustra el procedimiento paso a paso que se debe seguir para decidir sobre la presentación de una solicitud 510(k) para cambios de software en dispositivos existentes. En conclusión, la presente FDA describe en detalle el enfoque que deben seguir los Dispositivos Médicos a la hora de decidir si los cambios de software en un Dispositivos Médicos existente Dispositivos Médicos la presentación de un nuevo 510(k). Para obtener más información sobre la FDA , consulte a Freyr, un experto acreditado en materia de normativa. Manténgase informado. Mantenga el cumplimiento.









