2 min de lectura
Un «dispositivo predicado» es un Dispositivos Médicos ha sido previamente aprobado por la Administración US y MedicamentosUS FDA) y que ya se encuentra en el mercado, y que sirve como punto de referencia para los nuevos dispositivos médicos que solicitan la aprobación a través de la vía de autorización510(k) FDA.
Debe demostrarse que el producto en cuestión es al menos tan seguro y eficaz como el producto de referencia en cuanto a su uso previsto y sus características tecnológicas. Esta comparación se conoce como determinación de "equivalencia sustancial".
No es necesario que un nuevo dispositivo sea idéntico al dispositivo predicado para que sea sustancialmente equivalente a éste.
¿Cómo identificar un dispositivo predicado?
La base de datos FDAproporciona un código de producto de tres letras para cada clasificación de dispositivo. La base de datos FDA (k) contiene información sobre todos los dispositivos autorizados a través delproceso 510(k). Una vez que tenga el código de producto de tres letras, puede obtener una lista de todos los productos, todas las empresas y el nombre comercial de todos los competidores o posibles competidores que desee consultar. A continuación, puede realizar un análisis y una comparación en profundidad para reducir la búsqueda de un dispositivo predicado.
A continuación se muestra un diagrama de flujo que describe el proceso de identificación y reducción de la búsqueda de un dispositivo predicado.
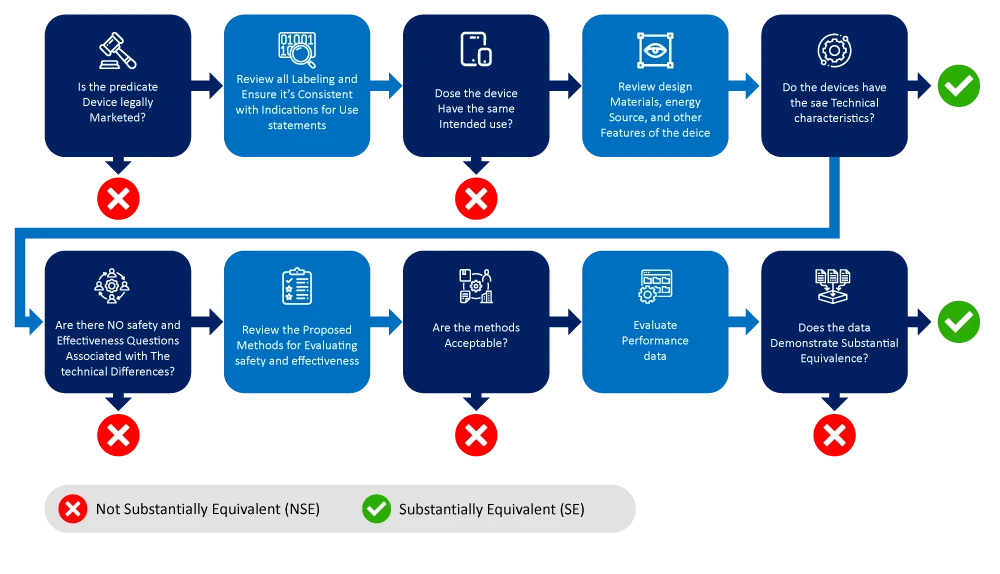
Factores a tener en cuenta para determinar el dispositivo o dispositivos de referencia
- Uso previsto: El uso previsto después del dispositivo predicado debe ser similar al del nuevo dispositivo. Por ejemplo, si el nuevo dispositivo está destinado a la monitorización cardíaca, el dispositivo predicado también debe ser un dispositivo de monitorización cardíaca.
- Características tecnológicas: El dispositivo predecesor debe ser idéntico al nuevo dispositivo en cuanto a características tecnológicas. Por ejemplo, el diseño, los materiales utilizados y el método de funcionamiento deben ser similares.
- Biocompatibilidad: Las evaluaciones de biocompatibilidad de un Dispositivos Médicos componente no deben limitarse a las materias primas utilizadas en el dispositivo y el proceso de fabricación, sino que también deben tenerse en cuenta los productos químicos adicionales. Sin embargo, este factor no se aplica a los dispositivos de diagnóstico in vitro (IVD).
- Última tecnología: El dispositivo predicado no debe ser anticuado y debe representar la última tecnología médica.
El dispositivo predicado es un factor clave para determinar si un nuevo Dispositivos Médicos comercializarse a través de la vía 510(k). Elegir el dispositivo predicado incorrecto podría dar lugar a un proceso de aprobación reglamentaria más costoso y lento, mientras que elegir el dispositivo predicado adecuado puede ayudar a reducir el coste y el tiempo necesarios para Dispositivos Médicos un nuevo Dispositivos Médicos . Si el dispositivo predicado no es adecuado, podría dar lugar a retrasos y gastos adicionales.
Si necesita ayuda con el proceso de presentación del formulario 510(k) de sus Dispositivos Médicos, programe una llamada con los expertos en normativa de Freyr, que le ayudarán a completar los trámites. Manténgase informado. Cumpla con la normativa.









