
3 min de lectura
Una notificación 510(k) o una notificación previa a la comercialización es una presentación realizada ante la Administración de Alimentos y Medicamentos de los Estados Unidos (US FDA) para demostrar que el dispositivo que se va a comercializar es seguro y eficaz, es decir, sustancialmente equivalente a un dispositivo comercializado legalmente o a un dispositivo predicado. A continuación se enumeran los tres (03) tipos de 510(k) que puede presentar un Dispositivos Médicos :
- Tradicional
- Abreviado
- 510(k) especial
En este blog, examinaremos los casos en los que su solicitud podría optar al segundo tipo, un 510(k) abreviado, según losFDA deFDA US
Una solicitud abreviada 510(k) se utiliza para demostrar la equivalencia sustancial con una norma, un control especial o una guía reconocidos mediante una declaración de conformidad (DoC). En una solicitud abreviada, los fabricantes demuestran la equivalencia sustancial con las normas reconocidas basándose en el uso de documentos de orientación o DoC, en lugar de un dispositivo predicado, para facilitar la revisiónFDA US A continuación se muestra el diagrama de flujo para determinar la equivalencia sustancial de una solicitud abreviada 510(k).
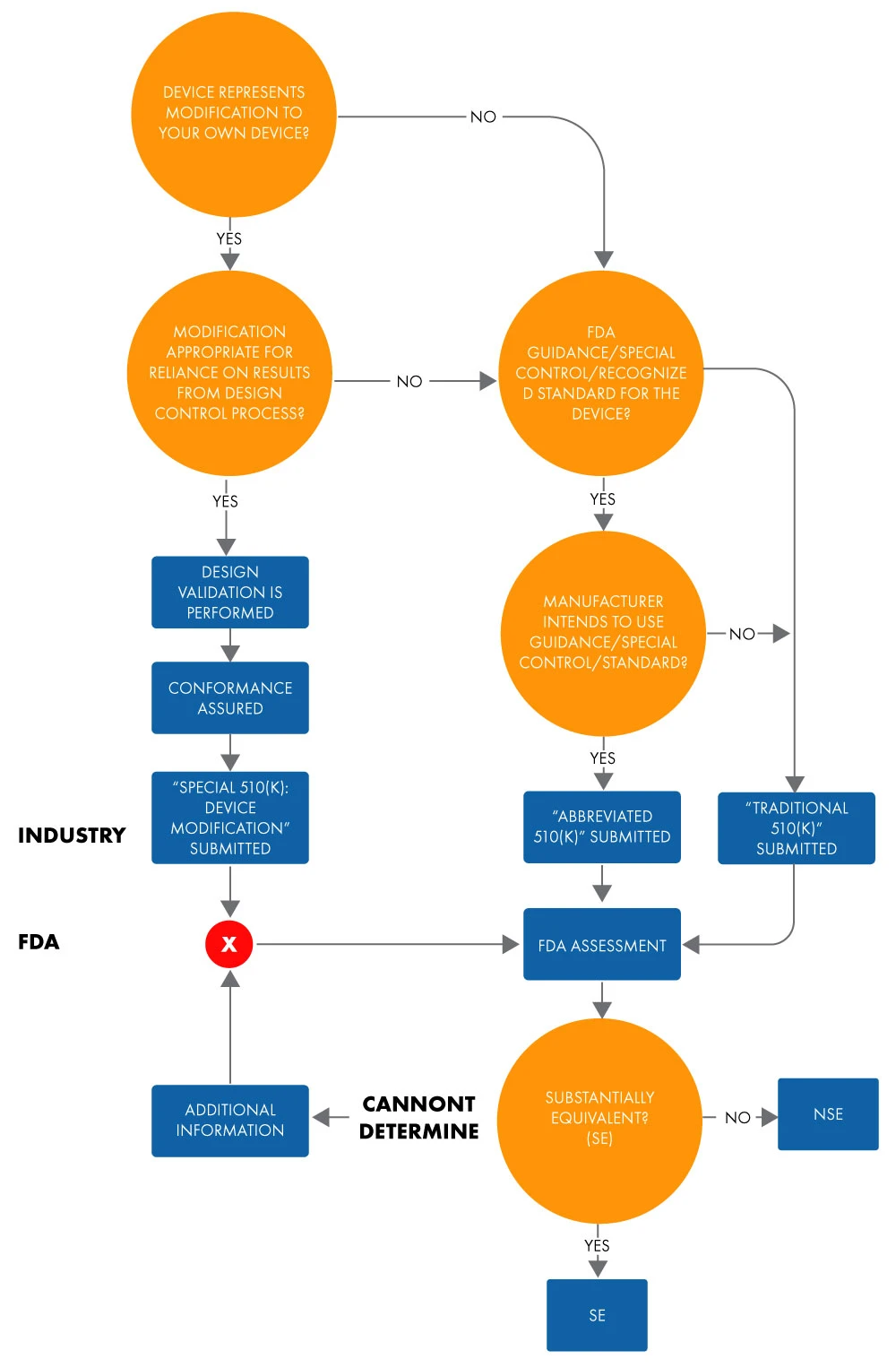
Figura 1: Intención de comercializar un dispositivo a través de 510(k)
El término "abreviado" sugiere que este tipo de proceso de aprobación 510(k) es más breve. Sin embargo, esto no es del todo cierto. Lleva el mismo tiempo que una aprobación 510(k) tradicional. Lo mismo ocurre con la documentación y los costes. Además, el formato de los 510(k) tradicionales y abreviados, en términos de capitulado y estructura, es similar.
Al presentar una solicitud 510(k) abreviada, debe basarse en los elementos identificados en 21 CFR 807.87 (solicitudes 510[k] tradicionales). Puede optar por presentar un 510(k) abreviado cuando la presentación se base en lo siguiente:
- DocumentosFDA : Alpresentar una solicitud abreviada 510(k), debe incluir un informe resumido que describa el cumplimiento del documento de orientación pertinente y cómo se utilizó durante el desarrollo y las pruebas del dispositivo.
- Demostración del cumplimiento de los controles especiales para el tipo de dispositivo: Debecumplir con controles especiales, tales como normas de rendimiento, vigilancia poscomercialización (PMS), registros de pacientes, desarrollo y difusión de directrices, recomendaciones, etc., que proporcionen una garantía razonable de la seguridad y eficacia del dispositivo. Una presentación abreviada 510(k) que se base en uno o varios controles especiales debe incluir lo siguiente. Un informe resumido que describa el cumplimiento de los controles especiales y cómo se utilizaron durante el desarrollo y las pruebas del dispositivo.
- Cómo se utilizaron los controles especiales para hacer frente a un riesgo o problema específico.
- Información que describa cualquier desviación de los controles específicos y los intentos del fabricante por cumplirlos.
- Norma(s) de consenso voluntario: Debe proporcionar una declaración de conformidad con la norma reconocida para una presentación abreviada 510(k) que se base en ella. La declaración de conformidad debe incluir lo siguiente:
- El nombre y la dirección del solicitante/patrocinador responsable del DoC.
- Detalles de la identificación del producto/dispositivo, incluidos los códigos de producto, el nombre comercial del dispositivo, el número de modelo y cualquier otro dato único de identificación del producto específico de la DdC en cuestión.
- Una declaración de conformidad.
- Una lista de las normas para las que es aplicable la DdC, incluida la opción u opciones seleccionadas para cada norma, en su caso.
- El númeroFDA US para cada norma.
- Fecha y lugar de expedición de la DdC.
- La firma, el nombre impreso y la función del patrocinador responsable de la DdC.
- Cualquier limitación de la validez de la declaración de conformidad (por ejemplo, durante cuánto tiempo es válida la declaración, qué se probó, concesiones hechas sobre los resultados de las pruebas, etc.).
En conclusión, un 510(k) abreviado es una forma útil para que los fabricantes de productos demuestren la equivalencia sustancial con normas reconocidas o controles especiales mediante una declaración de conformidad. Para poder optar a un 510(k) abreviado, los fabricantes de productos deben presentar un informe resumido en el que expliquen su adhesión a los documentos de orientación pertinentes, demostrar el cumplimiento de los controles especiales y presentar una declaración de conformidad con normas reconocidas. No obstante, es importante señalar que el proceso de aprobación, la documentación y el coste de un 510(k) abreviado son similares a los de un 510(k) tradicional.
¿Sus Dispositivos Médicos para una solicitud 510(k) abreviada? Si necesita ayuda para presentar su solicitud 510(k) abreviada, reach con nuestroexperto en normativa. ¡Manténgase informado! ¡Cumpla con la normativa!









