Visão geral do registo de dispositivos médicos nos EAU
Os Emirados Árabes Unidos (EAU), um importante país membro do CCG, possuem um sistema de saúde avançado. O seu potencial de mercado está comprovado e em constante crescimento, regido pelo Departamento de Controlo de Medicamentos do Ministério da Saúde e Prevenção (MOHAP). A governação centralizada e as barreiras linguísticas são os principais obstáculos ao registo de dispositivos médicos nos EAU, juntamente com as complexidades linguísticas e a falta de canais de comunicação eficientes com as autoridades de saúde.
![]()
Autoridade reguladora: Departamento de Controlo de Drogas do Ministério da Saúde e Prevenção (MOHAP)![]()
Regulamento: Orientações para o registo de dispositivos médicos nos EAU![]()
Via de regulação: Registo do produto![]()
Representante autorizado: É necessário um Representante Local Autorizado dos EAU![]()
Requisito do SGQ: ISO 13485:2016![]()
Avaliação dos dados técnicos: Comité de Registo dos Dispositivos Médicos![]()
Validade da licença: 5 anos![]()
Requisitos de rotulagem: Anexo 2 (2.5) do Guia de Registo de Dispositivos Médicos dos EAU![]()
Formato de apresentação: Papel![]()
Língua: Inglês
Classificação de Dispositivos Médicos dos EAU
Os EAU têm regras de classificação separadas para dispositivos médicos e IVD. As regras de classificação dos dispositivos médicos dos EAU estão em conformidade com as regras de classificação das diretivas da UE relativas aos dispositivos médicos. As classes de dispositivos de acordo com as regras de classificação dos EAU são as seguintes
| Critérios de risco | Classe de dispositivo médico |
|---|---|
| Baixo risco | I |
| Baixo Moderado Risco | IIa |
| Risco moderado - elevado | IIb |
| Risco elevado | III |
| Critérios de risco | Classe IVD |
|---|---|
| Baixo risco individual e baixo risco para a saúde pública | A |
Risco individual moderado e/ou Baixo risco para a saúde pública | B |
Risco individual elevado e/ou Risco moderado para a saúde pública | C |
| Risco individual elevado e risco elevado para a saúde pública | D |
Representante autorizado local dos EAU
Os fabricantes estrangeiros, sem escritório físico, devem nomear um representante local (LR) para atuar em seu nome. O representante local deve ser licenciado pelo Ministério da Saúde como armazém médico ou gabinete científico (no caso de gabinete científico, as actividades de importação e distribuição devem ser realizadas por um armazém médico licenciado nomeado). Os requerentes podem nomear o seu distribuidor como representante local. No entanto, a existência de um representante local independente, sem interesses comerciais, proporcionaria a flexibilidade necessária para nomear vários distribuidores nos EAU. Os dados do LR e do distribuidor devem ser fornecidos durante o registo do dispositivo.
Processo de classificação oficial com o MoHAP dos EAU
O MoHAP dos EAU introduziu um serviço de classificação oficial, particularmente útil quando não se tem a certeza se o seu produto necessita de registo. Este serviço classifica produtos de todos os tipos e formas com base na sua apresentação, composição, utilização e design. Os requisitos podem variar consoante a natureza do produto, a classe de risco e o estatuto regulamentar.
A carta de classificação indica se um produto tem de ser registado no MOHAP. Se o registo for necessário, o produto deve ser registado de acordo com a classe identificada na carta de classificação. Esta carta é válida por três anos a partir da data de emissão.
Os resultados da classificação oficial podem ser:
- Não necessita de registo no MOHAP
- Autorizado pelo MOHAP dos EAU como dispositivo médico, restrito a utilização profissional
- Autorizado pelo MOHAP dos EAU como dispositivo médico de venda livre
Registo de Dispositivos Médicos nos EAU
Certos dispositivos que não necessitam de registo do produto, nem de listagem ou aprovação prévia para a importação. Esses produtos isentos de registo ou listagem devem solicitar e obter uma licença de importação para serem comercializados nos EAU.
Relativamente a outros dispositivos, as importações não serão desalfandegadas a menos que seja emitida uma pré-aprovação para a importação da remessa pela DRCD. Esses dispositivos devem ser incluídos numa lista ou registados para importação para os EAU.
Listagem de dispositivos: Geralmente, os produtos utilizados em hospitais sob supervisão profissional e os dispositivos da classe I não são submetidos a uma avaliação pormenorizada e devem ser incluídos na lista. A agência emite um certificado de registo. Após a inclusão na lista, os dispositivos devem obter uma licença de importação para serem comercializados nos EAU.
Registo de dispositivos: A atividade de registo inclui o registo do local e do produto.
- Registo do local:O local de fabrico deve ser registado se o dispositivo fabricado nesse local for importado para os EAU pela primeira vez. Para os dispositivos subsequentes fabricados no mesmo local, bastará apenas o registo do dispositivo, não sendo necessário o registo do local.
- Registo do dispositivo:Estes dispositivos estão sujeitos a análise pelo comité técnico que, após aprovação, concederá um certificado de licença.
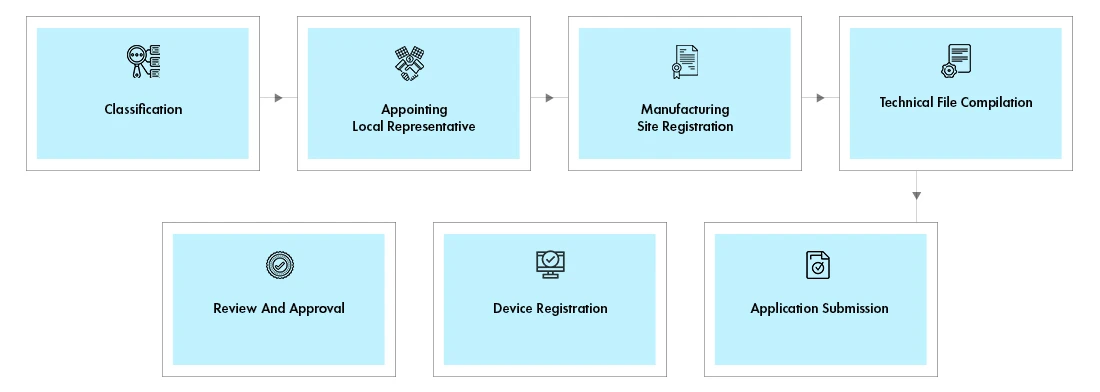
Fluxo do processo
Gestão do ciclo de vida do dispositivo pós-aprovação
- Gestão de alterações pós-aprovação - alterações às aprovações de Dispositivos Médicos existentes, tais como, adição de novas variantes, acessórios; adição de novas indicações de utilização, entre outras
- Manutenção das aprovações e do registo através do pagamento atempado das taxas administrativas e de registo
- Renovação de licenças
- Ligação entre o Ministério da Saúde e o fabricante
- Gestão das importações
Com um centro de entrega exclusivo no Dubai, Freyr detém uma posição de autoridade no mercado de Dispositivos Médicos dos EAU e descreve a classificação dos dispositivos para além de descodificar os regulamentos de orientação para uma melhor conformidade. Apoiamos os clientes na compilação de documentos de acordo com as normas e, assim, garantimos aprovações rápidas. Freyr oferece uma gama completa de serviços regulamentares relacionados com a comercialização bem sucedida de dispositivos.
Resumo
| Tipo de dispositivo | Listagem de dispositivos | Registo do dispositivo | Licença de importação |
|---|---|---|---|
Dispositivo isento de aprovação antes da importação (Conforme lista do Anexo 3) | NA | NA | SIM |
| SIM | NA | SIM |
| Todos os outros dispositivos | NA | SIM | SIM |
Especialização em Freyr
- Informações sobre regulamentação
- Diligência devida regulamentar
- Classificação formal de dispositivos médicos
- Registo do dispositivo
- Representação autorizada nos EAU
- Apoio à tradução
- Apoio à rotulagem
- Identificação e qualificação de distribuidores
- Gestão de alterações pós-aprovação
- Renovação e transferência de licenças
- Desembaraço aduaneiro