
Visão Geral da Aprovação Pré-Comercialização de Dispositivos Médicos da USFDA
O processo de Aprovação Pré-Comercialização (PMA) da USFDA é uma das vias de registo de dispositivos fornecidas pela US FDA, concebido principalmente para dispositivos médicos de Classe III da FDA. O processo de aprovação PMA da FDA para dispositivos de Classe III implica avaliações científicas e regulamentares meticulosas para avaliar a segurança e eficácia do dispositivo médico, garantindo que os mais altos padrões são cumpridos antes da autorização de introdução no mercado.
Marque uma reunião com os nossos especialistas em aprovação pré-comercialização
Quem deve submeter uma submissão de Aprovação Pré-Comercialização (PMA) de Dispositivos Médicos à USFDA?
Os fabricantes de dispositivos devem submeter uma submissão PMA se o dispositivo:
- É inovador.
- Pertence a uma classe de alto risco.
- Não pode ser encontrado na Base de Dados de Classificação de Produtos.
- Não é substancialmente equivalente (NSE) a dispositivos de Classe I, II ou III.
Obtenha aconselhamento especializado sobre a sua submissão de aprovação pré-comercialização
Qual é a diferença entre as submissões 510(k), PMA e De-Novo?
Aprovação Pré-Comercialização
- Dispositivo da Classe III que suporta a vida humana ou que apresenta um risco potencial e inaceitável de doença ou lesão.
- O processo de aprovação PMA da FDA requer ensaios clínicos.
- Requer inspeção no local antes de emitir a aprovação PMA.
- 180 dias de calendário
Classificação De Novo
- Dispositivos inovadores de Classe I e II que não possuem um dispositivo precedente válido.
- Requer dados de estudo clínico.
- Sem auditoria no local antes da aprovação De-Novo.
- 150 dias de calendário.
Registo 510(k)
- Dispositivos de Classe III da FDA que possuem equivalência substancial com o dispositivo de referência.
- Não requer testes em humanos.
- Sem auditoria no local antes da autorização 510(k).
- 90 dias de calendário.
Quais são os Diferentes Métodos de submissão para Aprovação Pré-Comercialização da FDA?
Os fabricantes podem optar por qualquer um dos quatro (04) métodos de submissão de PMA seguintes que melhor se adequem ao seu dispositivo:
- PMA Tradicional
- PMA Modular
- Protocolo de Desenvolvimento de Produto
- Isenção para Dispositivos Humanitários
Quais são os requisitos de dados para a aprovação pré-comercialização de dispositivos médicos?
De acordo com o 21 CFR parte 814, os requerentes devem submeter um formulário de submissão CDRH devidamente preenchido, os compromissos exigidos e um ficheiro técnico PMA bem elaborado à US FDA. O ficheiro técnico deve incluir os dados não clínicos e clínicos.
Dados Não Clínicos – Consiste em dados sobre microbiologia, toxicologia, imunologia, biocompatibilidade, stress, desgaste, prazo de validade e outros testes laboratoriais ou em animais.
Dados Clínicos – Consiste em dados sobre protocolos de estudo, dados de segurança e eficácia, reações adversas e complicações, falhas e substituições de dispositivos, informações do paciente, reclamações de pacientes, tabelas de dados de todos os indivíduos, resultados de análises estatísticas e qualquer outra informação das investigações clínicas.
Qual é o Processo de Submissão PMA?
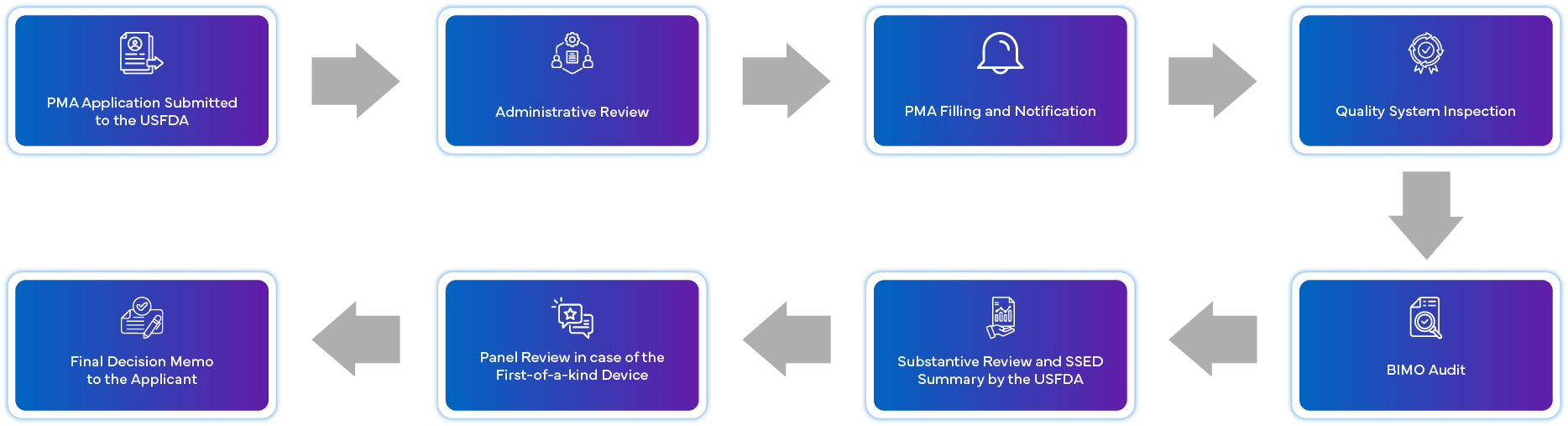
Quais são os Requisitos de Conformidade Pós-Aprovação para PMA?
Os dispositivos aprovados sob a via PMA devem cumprir os requisitos de pós-comercialização estabelecidos pela USFDA. O dispositivo deve cumprir o seguinte:
- Requisitos pós-aprovação impostos na ordem de aprovação PMA da FDA.
- Gestão de alterações pós-aprovação através da submissão atempada de suplementos de PMA relevantes
- Submissão para relatórios pós-aprovação (anuais)
- Regulamentos 21 CFR 803 para a Notificação de Dispositivos Médicos (MDR)
- Estudos de Vigilância Pós-Comercialização conforme exigido pela USFDA nas ordens de aprovação PMA.
Quais são as taxas da USFDA para a revisão da submissão PMA?
As taxas de utilizador MDUFA para PMA original e suplementos são as seguintes:
| Tipo de submissão | Taxas para o Ano Fiscal de 2023 (1º de outubro de 2022 a 30 de setembro de 2023) | |
|---|---|---|
| Taxa padrão | Taxa para pequenas empresas | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Suplemento de Acompanhamento de Painel | $353,238 | $88,309 |
| Suplemento de 180 Dias | $66,232 | $16,558 |
| Taxa anual para relatórios periódicos sobre um dispositivo de Classe III (PMAs, PDPs e PMRs) | $15,454 | $3,864 |
| Aviso de 30 Dias | $7,065 | $3,532 |
| Suplemento em Tempo Real | $30,908 | $7,727 |
Com experiência em lidar com submissões PMA, a Freyr pode ajudar a identificar e compilar as informações e auxiliar na preparação e revisão da submissão.
Experiência e Vantagens na Aprovação Pré-Comercialização de Dispositivos Médicos da USFDA
- Diligência Devida Regulatória
- Conformidade da Inspeção do Sistema de Qualidade
- Conformidade com a Auditoria BIMO
- Compilação de Dossier Técnico PMA
- Publicação e Criação de eCopy
- Validação e Submissão de eCopy
- Aborda a Resposta RTA e as Deficiências
- Serviços de Ligação até à Aprovação Pré-Comercialização da FDA
- Consulta para Deficiências
- Registo de Dispositivos e de Estabelecimentos
- Gestão de Suplementos PMA e Notificações de 30 dias
- Submissões anuais de relatórios periódicos
- Auditorias Simuladas e Formação sobre 21 CFR 820

- Experiência no tratamento de muitas submissões PMA da FDA para diversas categorias de dispositivos.
- Equipa especializada para a submissão de aprovação pré-comercialização da FDA de acordo com os requisitos regulamentares
- Suporte adicional para gerir questões relacionadas com PMA.
- Submissão atempada dos resultados
- Atualizado com as novas alterações da US FDA
