
Visão Geral dos Serviços de Conformidade com o EU MDR
O Regulamento dos Dispositivos Médicos da UE (MDR) entrou em vigor a 26 de maio de 2021, após o período de transição de 3 anos e uma extensão adicional de um ano devido à pandemia de COVID-19. Os dispositivos que estão a ser lançados no mercado da UE devem agora cumprir estes regulamentos e devem ser certificados CE de acordo com o EU MDR pelos Organismos Notificados acreditados ao abrigo destes regulamentos. Os dispositivos que já foram certificados CE de acordo com a Diretiva dos Dispositivos Médicos da UE (EU MDD), no entanto, têm períodos de carência antes de terem de cumprir integralmente os requisitos do EU MDR. Durante este período de carência, os dispositivos certificados ao abrigo tanto da EU MDD como do EU MDR coexistirão no mercado com o mesmo estatuto e sem serem sujeitos a discriminação. A Freyr oferece serviços de Conformidade com o EU MDR inigualáveis para ajudar as empresas de dispositivos médicos a cumprir os requisitos do EU MDR em tempo útil.
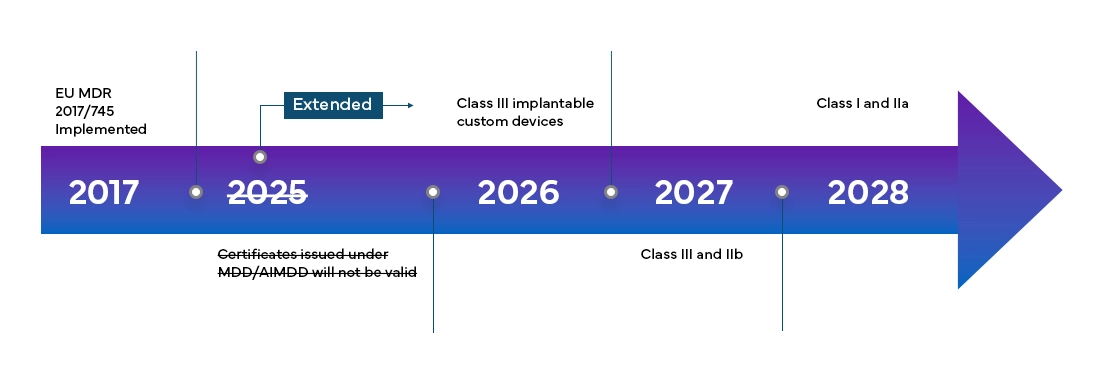
Cronograma de Transição e Novas Classificações de Dispositivos
O Regulamento Europeu dos Dispositivos Médicos (MDR) estará totalmente em vigor em todos os Member States da UE e nos Estados da Associação Europeia de Comércio Livre (EFTA) a partir de maio de 2021 e concede aos fabricantes um período de transição de 4 anos para a certificação completa do EU MDR.
O novo Regulamento Europeu dos Dispositivos Médicos (MDR), como se verificou, também trouxe alterações ao sistema de classificação de dispositivos existente, tais como:
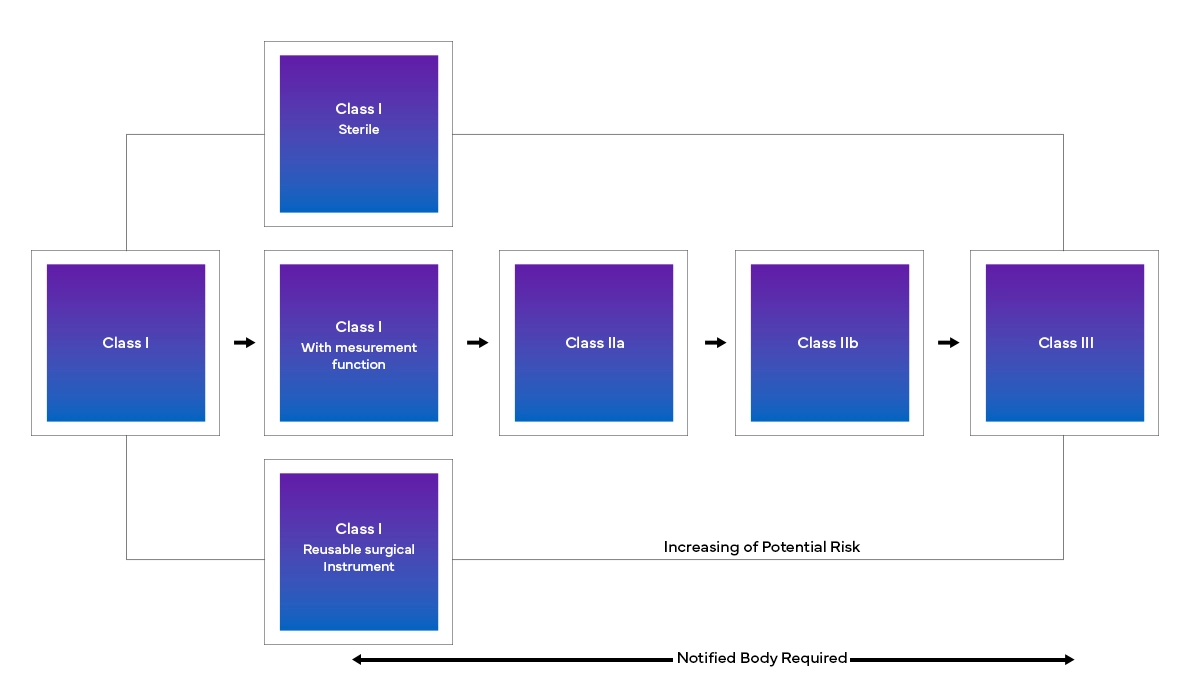
Desde a identificação das alterações exatas a serem feitas até à sua implementação em tempo real, os fabricantes podem ter de enfrentar uma série de desafios para cumprir os requisitos do EU MDR. Desde a descodificação da nova estrutura, a classificação precisa de um dispositivo, até à recolha e submissão de todos os dados, será necessária uma abordagem regulamentar mais detalhada e interfuncional para os fabricantes, para lidar com os novos Regulamentos Europeus dos Dispositivos Médicos. Com uma análise de lacunas rigorosa, a Freyr ajuda os clientes com o status quo e, assim, fornece a ação regulamentar necessária para a transição e conformidade com o EU MDR.
Obtenha aconselhamento especializado sobre a sua conformidade com o EU MDR
Serviços de Conformidade EU MDR
- Desenvolver uma estratégia clara de implementação do Regulamento dos Dispositivos Médicos (MDR)
- Compreender a nova legislação, realizar uma Análise de Lacunas aos atuais Sistemas de Gestão da Qualidade (SGQ) e processos existentes
- Desenvolver um plano detalhado com uma abordagem multifuncional para determinar os aspetos do sistema de qualidade que necessitarão de modificação, em conformidade com o novo Regulamento Europeu de Dispositivos Médicos
- Formar várias equipas para analisar o âmbito do produto, classificação, gestão do SGQ, etc., dentro da organização, com um único ponto de contacto em cada equipa
- Alocação e planeamento de recursos
- Considerar a interação do seu SGQ com outras regulamentações e aproveitar esta oportunidade para otimizar processos, permitindo flexibilidade para incorporar futuras alterações
- Analisar os dados de teste existentes e verificar quaisquer requisitos adicionais que o MDR estabelece
- Coordenar as expectativas e o plano de transição com os seus Organismos Notificados da UE
- Análise de Lacunas para Dispositivos Médicos existentes da Diretiva dos Dispositivos Médicos da UE (EU MDD) para os Regulamentos do EU MDR
- Suporte End-to-End para desenvolver o Relatório de Avaliação Clínica (CER), incluindo pesquisa bibliográfica de acordo com as diretrizes do Regulamento Europeu de Dispositivos Médicos (EU MDR).
- Serviços End-to-end para Relatórios de Vigilância Pós-Comercialização (PMSR), Relatório Periódico de Atualização de Segurança (PSUR) e Resumo da Segurança e Desempenho Clínico (SSCP)
- Aumento de Recursos Regulamentares com opções de implementação onshore e offshore
- Serviços de Representante Autorizado Europeu (RAE)
- Conformidade com o MDR e assistência na submissão aos Organismos Notificados
- Inteligência Regulamentar abrangendo o processo de importação de diferentes mercados regulamentados
- Conformidade com o SGQ e auditorias simuladas
- Sistema de Gestão Documental e ferramenta para empresas MDR
- Classificação e reclassificação de dispositivos de acordo com o risco
- Implementação e consultoria de UDI
- Serviços de Vigilância Pós-Comercialização em conformidade com o Regulamento dos Dispositivos Médicos da UE
- Consultoria em Gestão de Risco ISO 14971
- Formação interna e online
- Serviços e assistência de Pessoa Responsável pela Conformidade Regulamentar
- Identificação de Organismos Notificados MDR

Para suporte regulamentar End-to-End em EU MDR, contacte a Freyr







