Visão Geral dos Produtos Combinados de Dispositivos Médicos
No mundo dinâmico da saúde e inovação, os produtos combinados de dispositivos médicos tornaram-se uma ponte robusta que liga produtos farmacêuticos, dispositivos médicos e produtos biológicos. O mercado de produtos combinados está em rápido crescimento, com uma Taxa de Crescimento Anual Composta (CAGR) prevista de 8,9%, de 2023 a 2030. O setor de produtos combinados medicamento-dispositivo está preparado para um crescimento sustentado, impulsionado por avanços tecnológicos, infraestruturas de saúde melhoradas, rotas regulamentares mais fluidas, colaborações estratégicas e um compromisso com os cuidados centrados no paciente.
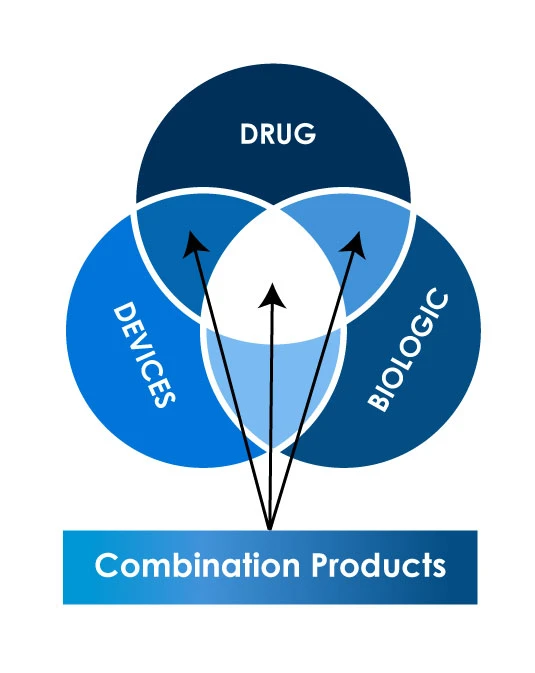
Diferentes Tipos de Produtos Combinados
Cenário Regulamentar Global para o Registo de Produtos Combinados
A interpretação do que constitui um produto combinado pode diferir de um país para outro, aumentando as complexidades do registo de tais produtos em vários países. Além disso, as exigências regulamentares e os procedimentos para produtos combinados podem apresentar variações na documentação, comunicação e validação. O panorama regulamentar para o registo de produtos combinados pode diferir significativamente em todo o mundo. Aqui estão as principais Autoridades Regulamentares que supervisionam globalmente estes dispositivos.
| País | Agência | Centros Responsáveis pela Aprovação |
|---|---|---|
| EUA | Gabinete de Produtos Combinados (OCP) | Centro de Avaliação e Investigação de Medicamentos (CDER) |
| Centro de Avaliação e Pesquisa de Produtos Biológicos (CBER) | ||
| Centro de Dispositivos e Saúde Radiológica (CDRH) | ||
| UE | Organismos Notificados (NBs) | Autoridade Competente Nacional (medicamentos) |
| Organismos Notificados (NBs) (dispositivos médicos) | ||
| Japão | Divisão de Avaliação e Licenciamento ou o Gabinete de Dispositivos Médicos/Produtos Celulares e à Base de Tecidos do Gabinete de Segurança Farmacêutica e Alimentar | Diretor da Divisão de Avaliação e Licenciamento (DMDL), Gabinete de Segurança Farmacêutica e Alimentar, Gabinete de Segurança Farmacêutica e Médica, Ministério da Saúde e Bem-Estar |
| China | Centro de Administração de Normalização de Dispositivos Médicos (CMDSA) | Centro de Avaliação de Dispositivos Médicos (CMDE) |
| Centro de Avaliação de Medicamentos (CDE) | ||
| Malásia | Agência Nacional de Regulamentação Farmacêutica | Agência Nacional de Regulamentação Farmacêutica (NPRA) |
| Agência de Dispositivos Médicos |
O registo de produtos combinados nos mercados internacionais necessita de uma abordagem personalizada, envolvendo uma estreita colaboração com as Agências de Saúde relevantes para aprovação. O processo típico para o registo de produtos combinados envolve os seguintes passos:
- Avaliar se um dispositivo específico cumpre os critérios para classificação como produto combinado.
- Categorizar os dispositivos com base nos riscos associados.
- Identificar as normas relevantes e os pré-requisitos de dados especificados pela respetiva Agência de Saúde.
- Gerar os dados necessários conforme exigido pela Agência.
- Compilar um ficheiro técnico de acordo com os requisitos específicos de cada país.
- Submeter a submissão e responder a quaisquer questões ou preocupações até que a aprovação seja obtida.
- Gerir o ciclo de vida do dispositivo após a aprovação.
As Nossas Competências
- Análise de risco inicial
- Pesquisa de mercado - Informações de mercado específicas do produto
- Reforço de equipa
- Elaboração da estratégia regulamentar
- Mercados e rotas potenciais
- Dossier de conceção e análise de risco
- Sistema de Gestão da Qualidade (SGQ) ISO 13485
- Programa de Auditoria Única de Dispositivos Médicos (MDSAP)
- Pré-avaliação QMS ISO 13485
- Estratégia regulamentar
- Freyr IMPACT (Plataforma de Informação Regulatória)
- Verificação e validação do design
- Gestão de riscos
- Elaboração da documentação técnica
- Estratégia regulamentar
- Requisitos regulamentares
- Ferramenta Freyr rDMS (Sistema de Gestão de Dados/Documentação)
- Validação de processo e clínica
- Rotulagem final e Artwork
- Representação no país
- Submissão regulamentar
- A marcação «Conformité Européenne» (CE) da União Europeia (UE) e a marcação de Avaliação de Conformidade do Reino Unido (UKCA)
- Certificação de acesso ao mercado global
- Apoio à auditoria de Organismos Notificados (ON)/Organismos Aprovados
- Representação no país
- Aprovações regulamentares
- Vigilância Pós-Comercialização (PMS)
- Acompanhamento Clínico Pós-Comercialização (PMCF)
- Manutenção anual do dossiê técnico (CER/Gestão de Risco)
- Renovações regulamentares
- Lançamentos em novos mercados
- Comunicação com a Autoridade Competente/Organismo Notificado/Organismo Aprovado
- Soluções de Farmacovigilância (FV) automatizada
Porquê a Freyr?
Registo de Dispositivos Médicos
- Estratégia regulamentar abrangente para produtos de combinação.
- Apoio regulamentar para documentos de desenvolvimento de produtos, como Dossiers de Histórico de Conceção (DHFs).
- Estratégia de conformidade com o Sistema de Gestão da Qualidade (SGQ).
- Conformidade regulamentar, análise de lacunas e remediação de documentos técnicos e sistemas de qualidade.
- Serviços de rotulagem regulamentar e redação técnica.
- Serviços de inteligência regulamentar e de mercado.
- Serviços de tradução de documentos e rotulagem.
- Serviços de contacto com Agências de Saúde.
- Serviços de artwork regulamentar.
- Serviços de Farmacovigilância e PMS.
- Serviços de publicação.
- Serviços de redação médica.

- Submissões bem-sucedidas para várias classes de IVDs.
- Pessoal dedicado e especializado para prestar apoio regulamentar a dispositivos médicos e IVDs.
- Entrega atempada dos resultados.
- Acesso a filiais locais para responder aos desafios da Autoridade e aos requisitos específicos de idioma.
- Apoio de representante local ou legal com um modelo de custo-eficácia.
- Serviços de gestão de recursos regulamentares/reforço de equipa.