
Conformidade com o registo de dispositivos médicos e panorama regulamentar na UE
A União Europeia representa um dos mercados de dispositivos médicos mais regulamentados e comercialmente atrativos do mundo. Juntamente com os 27 member states, os 3 EEA países e a Turquia, o mercado da União funciona ao abrigo de um ecossistema regulamentar harmonizado, definido pelo EU MDR (2017/745) para dispositivos médicos e pelo IVDR da UE (2017/746) para diagnósticos in vitro, garantindo a região normas uniformes de segurança e desempenho em todas as categorias de produtos. Para colocar legalmente um dispositivo no mercado da União Europeia, os fabricantes devem obter a marcação CE, que confirma a conformidade com o regulamento aplicável com base na classificação de risco do dispositivo e na via de avaliação da conformidade. O incumprimento destes requisitos pode resultar em consequências graves, incluindo a recolha do produto, a apreensão na alfândega, a suspensão da certificação ou a perda total do acesso ao mercado.
Ao abrigo do atual regime regulamentar:
- A marcação CE é obrigatória para todos os dispositivos médicos e dispositivos de diagnóstico in vitro (IVD) antes de poderem ser comercializados.
- O registo de dispositivos na EUDAMED está a ser implementado por fases, e vários módulos já se encontram ativos para utilização obrigatória.
- A nomeação de um Representante Autorizado Europeu (EAR) é obrigatória para qualquer fabricante sediado fora da UE/EEA/Turquia.
- As regras de classificação mais rigorosas do MDR/IVDR exigem documentação técnica sólida, evidências clínicas ou de desempenho e uma monitorização contínua ao longo do ciclo de vida.
A Freyr apoia os fabricantes ao longo de todo este percurso, facilitando uma entrada harmoniosa e em conformidade no mercado da União Europeia. Desde o desenvolvimento de uma estratégia de marcação CE e a elaboração da documentação técnica até à gestão dos pedidos junto dos Organismos Notificados e ao desempenho das funções de Representante Autorizado na UE, a Freyr garante a conformidade total com os regulamentos MDR/IVDR e um acesso sem obstáculos a todas as regiões da UE member states, EEA e à Turquia.
Processo passo a passo para a conformidade com a legislação da UE
O acesso ao mercado de um dispositivo médico e de um dispositivo de diagnóstico in vitro (IVD) no mercado da UE/União envolve várias etapas definidas. Eis como a Freyr gere todo o processo por si:
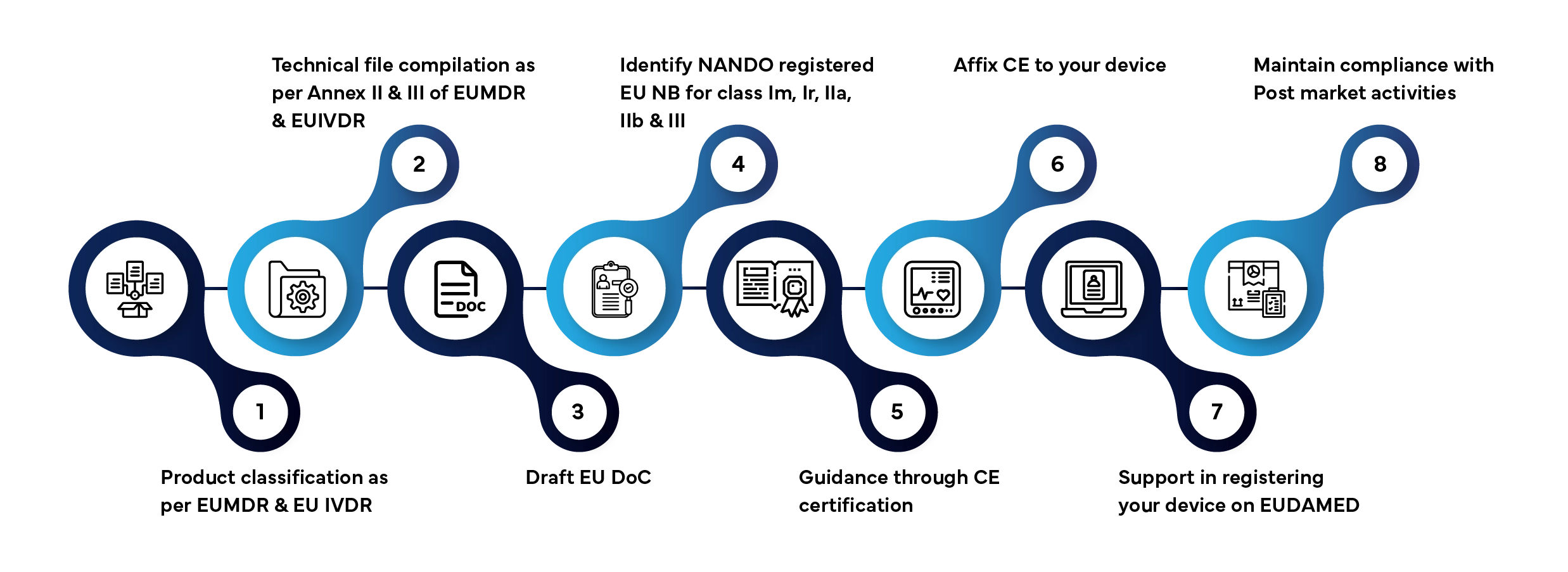
Prazo de processamento habitual: 3 a 12 meses, dependendo da classe do dispositivo e da disponibilidade da documentação.
Principais ofertas da Freyr Medical Device na UE
- Estratégia Regulamentar e Transição para o MDR/IVDR – Elaboramos planos de ação regulamentares « end-to-end » adaptados ao Regulamento sobre Dispositivos Médicos ( EU MDR ) e ao Regulamento da UE relativo aos Dispositivos de Diagnóstico In Vitro (IVDR), garantindo uma transição harmoniosa dos regimes das diretivas e o alinhamento com os requisitos atuais da UE.
- Documentação Técnica e Avaliação da Conformidade – A nossa equipa apoia o desenvolvimento, a revisão e a apresentação dos seus Dossiers Técnicos/Dossiers de Concepção, presta assistência nas relações com os Organismos Notificados e gere a marcação CE, o apoio aos testes de segurança e desempenho dos dispositivos e os processos de conformidade.
- Avaliação Clínica/de Desempenho: A Freyr oferece serviços especializados na elaboração de CERs, PERs, planos de « PMCF »/PMPF, PSURs, SVR, CPR para dispositivos de diagnóstico in vitro (IVDs), documentação de risco e conteúdos de avaliação biológica, garantindo clareza técnica e rigor regulamentar em todas as classes de dispositivos.
- Apoio ao registo no UDI e no EUDAMED – Asseguramos que o seu sistema de Identificação Única de Dispositivos (UDI) está em conformidade e prestamos assistência no registo na base de dados europeia de dispositivos EUDAMED e na gestão do ciclo de vida associada.
- Representante Autorizado Europeu (EAR) e Representação Local – Para fabricantes fora da UE/EEA/Turquia, atuamos como o seu Representante Autorizado Europeu (EAR) mandatado e prestamos apoio local em matéria de conformidade em toda a UE member states.
- Vigilância pós-comercialização (PMS) – A Freyr apoia-o no estabelecimento, implementação e manutenção de sistemas de PMS, incluindo o Plano de Vigilância Pós-Comercialização (PMSP), Relatório de Vigilância Pós-Comercialização (PMSR), Relatório Periódico de Atualização de Segurança (PSUR), planos de Acompanhamento Clínico/de Desempenho Pós-Comercialização (PMCF/PMPF), relatórios de vigilância, FSCAs, FSNs e CAPAs, garantindo a manutenção contínua da marcação CE e o acesso sustentável ao mercado, em conformidade com o ciclo de vida do produto.
- Registo de dispositivos com marcação CE: A Freyr gere todo o processo de registo da marcação CE na UE, prestando apoio na avaliação da conformidade, elaborando dossiês em conformidade, colaborando com os organismos notificados e garantindo aprovações atempadas em todas as categorias de dispositivos médicos e de diagnóstico in vitro (IVD).
- Apoio ao SGQ: Prestamos apoio à implementação e manutenção de Sistemas de Gestão da Qualidade em conformidade com a norma ISO 13485, alinhados com os requisitos de qualidade e segurança da Diretiva EU MDR/IVDR e com as expectativas dos organismos notificados.
- Conformidade com os requisitos de rotulagem: A nossa equipa garante que a sua rotulagem, instruções de utilização, embalagem e símbolos cumprem os requisitos do MDR/IVDR e os requisitos linguísticos multilingues da UE, mantendo a consistência e a conformidade nos 27 Estados-Membros da UE member states.
Oferta de serviços do Representante Autorizado na UE (EAR)

Registo de dispositivos junto das autoridades da UE
No que diz respeito aos fabricantes não pertencentes à UE, a Freyr atua como Representante Autorizado na UE (EAR) nomeado, em conformidade com os requisitos do MDR/IVDR. Enquanto entidade sediada na Alemanha, a Freyr presta apoio às atividades de registo de dispositivos junto da autoridade competente alemã relevante e mantém os registos regulamentares exigidos. No que diz respeito a outros Member States da UE, a Freyr presta apoio regulamentar, conforme aplicável, para ajudar a garantir a conformidade dos dispositivos e facilitar a sua colocação no mercado da União.

Documentação e Garantia de Conformidade
Os nossos especialistas em regulamentação verificam se a sua Declaração de Conformidade (DoC), os Certificados CE e os Dossiers Técnicos estão completos, atualizados e em conformidade com o MDR/IVDR. A Freyr garante que tudo está totalmente preparado para a avaliação da conformidade e a apresentação dos pedidos de marcação CE.

Resposta a pedidos de informação das autoridades competentes
Se necessário, a Freyr trata, em seu nome, de toda a comunicação direta e dos pedidos de esclarecimento por parte das autoridades competentes da UE ou dos organismos notificados, garantindo respostas atempadas e precisas e ajudando a evitar atrasos nas aprovações ou nas revisões regulamentares pós-comercialização.

Vigilância e comunicação de incidentes
Na qualidade de seu EAR, a Freyr atua como principal ponto de contacto para as comunicações relacionadas com a segurança. Sempre que necessário, coordenamos as notificações de incidentes, as Ações Corretivas de Segurança no Terreno (FSCA) e os relatórios de vigilância entre o fabricante, os profissionais de saúde e as autoridades, de modo a garantir uma resposta adequada e o cumprimento da regulamentação.

Preparação para inspeções e auditorias
A Freyr mantém toda a documentação, correspondência e registos obrigatórios para auditorias e inspeções das autoridades. A nossa equipa assegura que a documentação técnica, a rotulagem e os registos pós-comercialização estejam prontamente disponíveis e em total conformidade com os requisitos do MDR/IVDR.
Porquê colaborar com a Freyr?
- End-to-end conhecimento especializado em matéria de regulamentação que abrange desde o registo pré-comercialização até à vigilância pós-comercialização, gerindo todas as fases do cumprimento normativo.
- Um historial comprovado, com mais de 2000 registos de dispositivos concluídos com sucesso em diversas categorias.
- Forte presença na UE, com sede na Alemanha (EAR), complementada por especialistas em regulamentação no terreno em Reading e apoiada pelas equipas globais de execução da Freyr.
- Planeamento de transição personalizado que proporciona apoio estratégico para obter a certificação CE de forma harmoniosa e económica.
- Equipas de entrega multirregionais com apoio no terreno através das operações da Freyr na UE, sediadas na Alemanha.
- Experiência comprovada na implementação de medidas corretivas no âmbito do MDR/IVDR e na transição de dispositivos antigos
- Gestão transparente do projeto com comunicação direta com o Organismo Notificado

Celebrando o sucesso dos clientes
Dispositivos Médicos
Apoio ao Registo e LR
Global
A Freyr tem sido um parceiro indispensável para alcançar uma rápida escalabilidade global para o nosso negócio de Software como Dispositivo Médico (SaMD). Como startup, adquirir experiência em regulamentações mundiais é proibitivo em termos de custos. Os preços competitivos e os serviços personalizados da Freyr permitiram-nos obter essa experiência a uma fração do custo de recursos a tempo inteiro. A recetividade e adaptabilidade da sua equipa às prioridades do projeto facilitaram grandemente o nosso progresso. Recomendamos a Freyr a qualquer empresa que procure orientação e apoio especializados no domínio regulamentar de dispositivos médicos.
Arie Henkin
VP - Qualidade e Regulamentação, com sede na Austrália, Empresa Líder de SaMD
Dispositivos Médicos
Serviços de Representação Suíça
Japão e Suíça
Gosto genuinamente de trabalhar com a Freyr e considero-os um ativo verdadeiramente valioso e uma extensão da minha própria equipa. São fiáveis e precisos, e os seus preços são competitivos. Além disso, não hesitarei em colaborar novamente com a Freyr.
Darren Mansell
Gestor de Assuntos Regulamentares, com sede no Reino Unido, Empresa Global de Design e Fabrico de Dispositivos Médicos
Dispositivos Médicos
Serviços de Registo e AR
Malásia e Indonésia
A Freyr oferece um serviço fiável com experiência em muitos países. Posso confiar na Freyr para fornecer as informações necessárias para tomar uma decisão informada antes de celebrar um acordo formal de âmbito de trabalho. Uma vez iniciado um projeto, a equipa Freyr age profissionalmente para executar o trabalho com excelente comunicação do progresso.







