Garantir a conformidade sem falhas com os regulamentos e o registo de dispositivos médicos do Reino Unido
Os fabricantes estrangeiros têm de cumprir o Regulamento do Reino Unido relativo aos Dispositivos Médicos de 2002 (na sua versão alterada). Todos os dispositivos médicos e dispositivos de diagnóstico in vitro (IVD) têm de ser registados junto da Agência de Regulamentação dos Medicamentos e Produtos de Saúde ( MHRA ) antes da sua comercialização no Reino Unido. Os fabricantes não britânicos são obrigados a nomear um Responsável no Reino Unido (UKRP) para os representar. O Regulamento do Reino Unido relativo aos Dispositivos Médicos de 2002 ( MHRA ) estabelece requisitos rigorosos em matéria de documentação, rotulagem e comunicação de vigilância, a fim de garantir a segurança dos doentes e a rastreabilidade dos produtos.
Orientar-se entre os novos requisitos propostos no resultado da consulta para o Reino Unido pode revelar-se complexo no que diz respeito à marcação UKCA, aos requisitos em matéria de dados e às expectativas pós-comercialização. Muitas empresas enfrentam desafios relacionados com dossiers técnicos incompletos, classificações divergentes dos dispositivos e um conhecimento limitado do Sistema de Registo de Dispositivos Online (DORS). As atualizações regulamentares e a evolução das orientações da MHRA complicam ainda mais o planeamento e a afetação de recursos.
A Freyr simplifica todas as etapas da entrada no mercado do Reino Unido. Tratamos dos pedidos de DORS, atuamos como o seu « UKRP », garantimos que a documentação esteja pronta e colmatamos as lacunas de conformidade durante a transição da marcação CE para a UKCA. Os nossos especialistas em regulamentação orientam-no através das políticas em constante mudança da « MHRA », ajudando-o a obter um acesso ao mercado em conformidade, atempado e económico.
Processo passo a passo de registo de dispositivos médicos no Reino Unido
O registo de um dispositivo médico no Reino Unido envolve várias etapas definidas. Eis como a Freyr gere todo o processo por si:
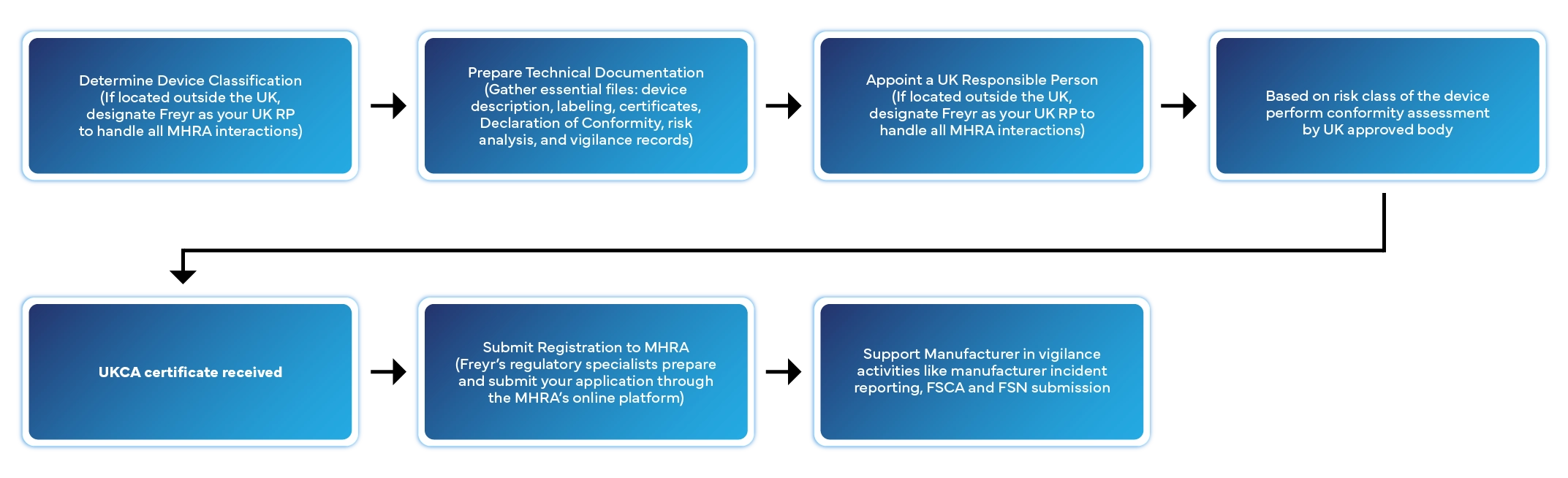
Prazo de processamento habitual: 2 a 6 semanas, dependendo da classe do dispositivo e da completude da documentação.
Principais ofertas da Freyr Medical Device UK

Registo de dispositivos A Freyr gere todo o processo de registo nos Estados Unidos da América ( MHRA ) através da plataforma DORS, garantindo submissões em conformidade, dados precisos e aprovações atempadas para todas as categorias de dispositivos.

Responsável no Reino Unido Na qualidade de seu « UKRP » designado, a Freyr representa-o perante a « MHRA », trata de toda a comunicação e assegura a conformidade contínua e a monitorização da vigilância.

TF / Compilação de dossiês A Freyr compila ficheiros técnicos e dossiês que cumprem os requisitos regulamentares do Reino Unido para a marcação UKCA, garantindo a preparação para auditorias, inspeções e apresentações.

Apoio ao SGQ Prestamos assistência na implementação e manutenção de Sistemas de Gestão da Qualidade em conformidade com a norma « ISO 13485 », adaptados às exigências do MDR do Reino Unido e da norma « MHRA ».

Apoio à Redação de Documentação Regulamentar A Freyr oferece serviços especializados de elaboração de CERs, planos de PMS, PSURs e documentação de gestão de riscos, garantindo clareza técnica e rigor regulamentar.

Rotulagem e Conformidade A nossa equipa garante que a sua rotulagem, as instruções de utilização e as embalagens cumprem os requisitos de rotulagem e linguísticos da certificação UKCA, mantendo a consistência e a conformidade.

Vigilância pós-comercialização A Freyr apoia as atividades de vigilância pós-comercialização (PMS), incluindo a notificação de eventos adversos, a apresentação de relatórios de vigilância e as atualizações do « MHRA », com vista a garantir a manutenção do acesso ao mercado.
Pessoa Responsável da Freyr UK (UKRP) Oferta de Serviços
A Freyr atua como o seu « UKRP » (Responsável pelo Mercado do Reino Unido) designado, garantindo o cumprimento integral do Regulamento de 2002 relativo aos Dispositivos Médicos do Reino Unido, disponível em MHRA. Representamos a sua empresa a nível local no Reino Unido e tratamos de toda a comunicação com a Autoridade de Regulamentação dos Medicamentos e Produtos de Saúde ( MHRA).
- Registo de dispositivos no site MHRA
A Freyr atua como a sua Pessoa Responsável designada no Reino Unido (UKRP) para gerir todo o processo de registo no site MHRA através da plataforma DORS, garantindo que cada dispositivo seja corretamente registado, verificado e elegível para venda no Reino Unido. - Documentação e Garantia de Conformidade
Os nossos especialistas em regulamentação asseguram que a sua Declaração de Conformidade, documentação técnica e certificações de produto estejam disponíveis e mantêm uma cópia para disponibilizar a documentação a MHRA, mediante pedido. - Resposta a consultas d MHRA
A Freyr trata diretamente de todas as comunicações e esclarecimentos com a MHRA em seu nome, garantindo respostas atempadas e precisas a consultas regulamentares ou análises pós-comercialização. - Vigilância e Comunicação de Incidentes
Na qualidade de seu « UKRP », a Freyr atua como principal ponto de contacto para questões relacionadas com a segurança. Coordenamos a comunicação entre fabricantes, profissionais de saúde, doentes e a « MHRA » relativamente a eventos adversos, garantindo a comunicação adequada e a implementação de ações corretivas. - Preparação para inspeções e auditorias
A Freyr mantém toda a documentação e correspondência necessárias para as inspeções e auditorias da MHRA . A nossa equipa garante que os ficheiros técnicos, a rotulagem e os registos pós-comercialização estejam prontamente disponíveis.
Agende uma reunião com os nossos especialistas hoje
Porquê colaborar com a Freyr?
- End-to-end conhecimento especializado em matéria de regulamentação que abrange desde o registo pré-comercialização até à vigilância pós-comercialização, gerindo todas as fases do cumprimento normativo.
- Um historial comprovado, com mais de 1 500 registos de dispositivos concluídos com sucesso em diversas categorias.
- Presença local no Reino Unido, com especialistas em regulamentação no terreno em Reading, apoiados por equipas de prestação de serviços a nível global.
- Um plano de transição personalizado que oferece apoio estratégico para a transição da certificação CE para a UKCA de forma harmoniosa e económica.
- Comunicação transparente através do contacto direto com MHRA e atualizações proativas sobre conformidade aos clientes.
- Com a confiança de marcas globais, a Freyr presta serviços a mais de 470 fabricantes em todo o mundo como seu parceiro em matéria de conformidade regulamentar.
Perguntas Frequentes (FAQs)
01. Em que consiste o processo de registo de dispositivos médicos no Reino Unido?
O processo de registo de dispositivos médicos no Reino Unido implica a notificação à Agência Reguladora de Medicamentos e Produtos de Saúde ( MHRA , MHRA) relativamente ao seu dispositivo antes de este ser comercializado na Grã-Bretanha. Os fabricantes devem fornecer os dados da empresa, a classificação do dispositivo e a documentação técnica. Os fabricantes não sediados no Reino Unido devem também nomear um Responsável no Reino Unido (UK RP) para tratar do registo e das comunicações relativas à conformidade contínua.
02. Quem tem de nomear um Responsável no Reino Unido (UK RP)?
Qualquer fabricante sediado fora do Reino Unido deve nomear um Responsável no Reino Unido (UK RP) antes de colocar dispositivos no mercado britânico. O UK RP atua como ponto de contacto regulamentar do fabricante, garantindo que toda a documentação técnica, declarações e comunicações com a Agência de Medicamentos e Produtos de Saúde do Reino Unido ( MHRA ) sejam devidamente mantidas. Os fabricantes sediados no Reino Unido podem interagir diretamente com a Agência de Medicamentos e Produtos de Saúde do Reino Unido ( MHRA ) sem necessidade de um UK RP.
03. Que documentação é necessária para o registo n MHRA ?
A Lei de Produtos Médicos ( MHRA ) exige documentação essencial, incluindo a Declaração de Conformidade, a descrição e classificação do dispositivo, os dados do fabricante e as informações de rotulagem. No caso de dispositivos de maior risco, os ficheiros técnicos e as evidências clínicas também podem ser analisados. Dispor de documentação completa e pronta para auditoria garante aprovações mais rápidas e inspeções pós-comercialização mais tranquilas por parte da Agência de Controlo de Alimentos e Medicamentos ( MHRA ) ou dos representantes autorizados.
04. Quanto tempo demora o processo de registo no « MHRA »?
Normalmente, o registo no âmbito da « MHRA » demora entre 2 a 6 semanas, dependendo da classe do dispositivo, da completude da documentação e da eventual participação de um Responsável no Reino Unido. Os dispositivos simples da Classe I podem ser processados mais rapidamente, enquanto os produtos complexos ou de maior risco podem demorar mais tempo devido à validação adicional de dados e às revisões da documentação técnica.
05. Qual é a diferença entre a marcação CE e a marcação UKCA?
A marcação CE atesta a conformidade com os regulamentos da União Europeia, enquanto a marcação UKCA (UK Conformity Assessed) aplica-se aos dispositivos comercializados na Grã-Bretanha. Desde o Brexit, a marcação UKCA substituiu a CE no mercado do Reino Unido, embora a marcação CE continue a ser aceite a título temporário. A Irlanda do Norte continua a reconhecer as marcações CE e CE UKNI ao abrigo das regras de alinhamento com a UE.
06. Quais são as responsabilidades pós-comercialização após o registo?
Após o registo, os fabricantes e os responsáveis no Reino Unido devem realizar a vigilância pós-comercialização, comunicar incidentes adversos, atualizar o site MHRA sobre alterações ao produto ou à rotulagem e renovar os registos, conforme necessário. Devem também conservar a documentação técnica durante, pelo menos, 10 anos. Estas atividades garantem a conformidade contínua e salvaguardam a saúde dos doentes ao longo de todo o ciclo de vida do dispositivo.
07. Com que frequência devem ser atualizadas as informações de registo d MHRA ?
As informações de registo devem ser atualizadas imediatamente sempre que houver uma alteração na classificação do dispositivo, na rotulagem, no local de fabrico ou na entidade responsável. A Agência de Medicamentos e Produtos de Saúde do Reino Unido ( MHRA ) espera que a Pessoa Responsável no Reino Unido ou o fabricante procedam a atualizações imediatas, a fim de manter registos de mercado precisos. As auditorias internas regulares ajudam a garantir que os seus dados se mantêm atualizados e em conformidade com a evolução da regulamentação.
08. Quais são as sanções aplicáveis em caso de incumprimento dos regulamentos d MHRA ?
O incumprimento dos requisitos de registo ou pós-comercialização de dispositivos médicos no Reino Unido pode resultar em advertências regulamentares, retirada do produto do mercado, multas ou ações judiciais. A Agência de Medicamentos e Produtos de Saúde ( MHRA ) também tem competência para suspender ou revogar a autorização de comercialização. Uma abordagem proativa em matéria de conformidade e a nomeação de um Responsável qualificado no Reino Unido ajudam os fabricantes a evitar sanções e a manter a continuidade da comercialização.
09. De que forma é que a Freyr simplifica o registo de dispositivos médicos no Reino Unido?
A Freyr presta apoio no âmbito d end-to-end , desde a função de Pessoa Responsável no Reino Unido (UKRP) até ao tratamento do registo no âmbito d MHRA , da documentação técnica e da conformidade contínua. Os nossos consultores experientes simplificam o processo, reduzem os prazos de aprovação e ajudam a garantir o acesso contínuo ao mercado.