SaMD nos US Visão geral
Respondido nesta página
- O meu software é um SaMD?
- Que classe de risco de SaMD requer um 510(k)?
- Registo e conformidade SaMD - o processo 510(k)
- Durante quanto tempo é válida a autorização?
O meu software é um SaMD?
De acordo com o Fórum Internacional de Reguladores de Dispositivos Médicos (IMDRF), SaMD é uma:
- Software destinado a ser utilizado para um ou mais fins médicos.
- Realiza estes objectivos sem fazer parte de um dispositivo médico de hardware.
Que classe de risco de SaMD requer um 510(k)?
Determinar a SaMD para o seu software é um passo importante no processo de registo. Depois que o seu software for classificado como SaMD, é essencial compreender o caminho regulatório necessário para entrar no mercado dos US. SaMD normalmente categorizado em diferentes classes, com base nos seus níveis de risco. SaMD Classe II SaMD considerado de risco moderado, requer uma autorização 510(k) e depende da comprovação de equivalência substancial com o dispositivo predicado comercializado legalmente. O processo de autorização garante que SaMD seu SaMD substancialmente equivalente aos dispositivos existentes, o que ajuda a garantir a sua segurança e eficácia antes de ser comercializado.
Registo e conformidade SaMD - o processo 510(k)
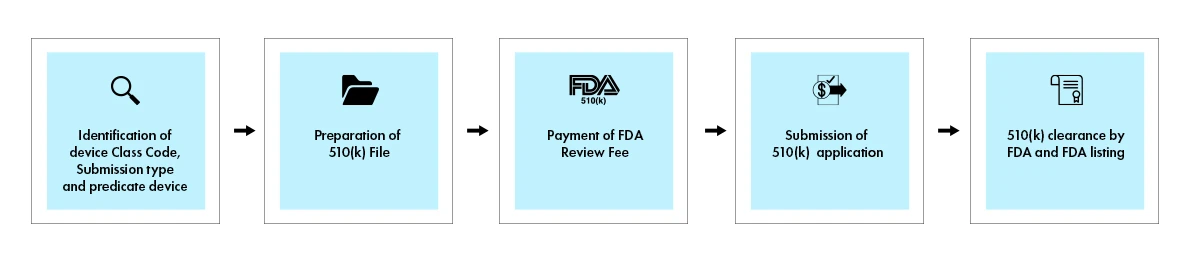
O processo 510(k) envolve uma submissão abrangente que demonstra equivalência substancial a um dispositivo predicado comercializado legalmente. Quando uma decisão é tomada, a US and Drug Administration (FDA) US emite uma carta de decisão ao submetente por e-mail. Uma submissão 510(k) submissão recebe uma carta de decisão de equivalência substancial é considerada «aprovada». Em seguida, é listada na base de dados 510(k), com o resumo 510(k) em anexo. A figura abaixo fornece uma visão geral das principais etapas envolvidas no processo 510(k).
Durante quanto tempo é válida a autorização?
Uma autorização 510(k) permanece válida até que sejam feitas alterações significativas no dispositivo ou nos regulamentos aplicáveis. No entanto, é importante observar que aFDA US FDA solicitar relatórios periódicos ou informações adicionais para garantir a conformidade e a segurança contínuas.
Em conclusão, o registo da SaMD requer um conhecimento profundo da classificação SaMD , da conformidade com a SaMD e dos processos regulamentares. A procura de serviços de consultoria SaMD pode fornecer orientação especializada para navegar pelas complexidades e garantir um resultado de registo bem-sucedido.
SaMD de SaMD nos US
- EstratégiaFDA abrangenteFDA US para SaMD.
- Classificação SaMD .
- Identificação do dispositivo predicado.
- Estabelecer a equivalência substancial com o dispositivo original.
- Análise de lacunas paraFDA US .
- Compilação do arquivo técnico 510(k), de acordo com as Orientações para Submissões Pré-comercialização de SoftwareFDA US
- Criação do modelo eCopy/eSTAR.
- Validação e apresentação do modelo eCopy/eSTAR.
- Serviços de ligação para a aprovação de dispositivos.
- Apoio à resposta da RTA e às deficiências da AINN.
- Serviços de consultoria para resolver deficiências.
- Registo do estabelecimento junto daFDA US .
- Listagem de dispositivos e manutenção da base de dados FURLS.
- Serviços do Representante Legal (RL).

- Vasta experiência com diversos registos 510(k).
- Experiência com compilação 510(k), de acordo com os requisitos de Notificação Pré-comercialização (510[k])FDA US .
- Apoio adicional para o tratamento de consultas 510(k).
- Apresentação atempada dos resultados.
- Em dia com as novas alteraçõesFDA US sobre SaMD.





