Declaração de Trabalho (SOW) para Dispositivo Médico Ativo e Dispositivo Médico Não Ativo 510(K) Descrição Geral da Apresentação
Na Freyr, a nossa equipa de especialistas compila e destila diligentemente as informações mais recentes essenciais para as suas submissões 510(K), abrangendo dispositivos médicos activos e não activos. Isto garante que possui os conhecimentos necessários para navegar com confiança no quadro regulamentar. Desde o esclarecimento dos contrastes entre dispositivos activos e não activos até ao aprofundamento dos meandros da submissão 510(K), selecionámos um extenso repositório de recursos para sua referência principal. Embarque numa viagem para dominar a submissão 510(K) de dispositivos médicos activos e a submissão 510(K) de dispositivos médicos não activos com o nosso guia com tudo incluído.
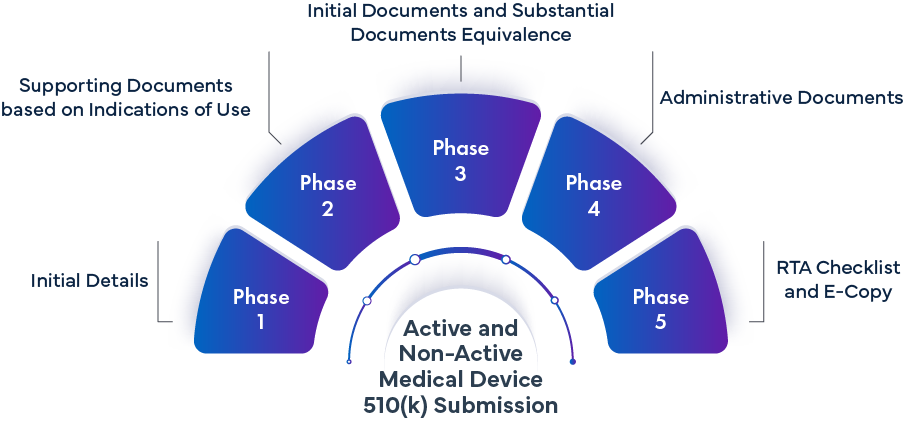
Fase -1 Pormenores iniciais | ||
|---|---|---|
Requisitos | Âmbito de aplicação do requerente 510(k) | Âmbito de aplicação de Freyr |
| Utilização prevista |
|
|
| Declaração de Indicações de Utilização (Formulário 3881) |
|
|
| Descrição do dispositivo |
|
|
| Normas e orientações |
|
|
| Dispositivo de predicado |
|
|
| Resumo 510(K) |
|
|
Fase 2: Documentação de apoio baseada nas indicações de utilização | |||
|---|---|---|---|
Requisitos do documento | Âmbito de aplicação do requerente 510(k) | Âmbito de aplicação de Freyr | |
| 2.1 | Desenho do dispositivo | Apresentar o ficheiro de desenho do dispositivo para garantir uma representação exacta da conceção do dispositivo | Iniciar um pedido formal para um Desenho de Dispositivo de um dispositivo Ativo. Rever minuciosamente e documentar meticulosamente as informações necessárias para a apresentação de 510(k). |
| 2.2 | Conceção e desenvolvimento do dispositivo | Apresentar o ficheiro de conceção e desenvolvimento do dispositivo ativo, incluindo todas as informações e documentação relevantes. | Apresentar um pedido de conceção e desenvolvimento de um dispositivo ativo. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.3 | Ficha de dados de segurança do material | Fornecer as fichasMSDS dados de segurança dos componentes essenciais do dispositivo ativo, garantindo informações completas sobre a sua segurança e composição | Enviar uma requisição de uma ficha de dados de segurança dos componentes essenciais do dispositivo ativo. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.4 | Fluxograma de fabrico | Fornecer um fluxograma de fabrico que descreva pormenorizadamente o processo de produção do dispositivo ativo, fornecendo uma representação visual das etapas de fabrico e da respectiva sequência | Apresentar um pedido de Ficha de Dados de Segurança dos MateriaisMSDS) dos componentes essenciais do dispositivo ativo. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.5 | Descrição do dispositivo | Fornecer pormenores completos, incluindo: o Uma visão geral do dispositivo o Funções e modos de funcionamento o Diagramas de blocos o Fotografias, cabos e acessórios relevantes o Interoperabilidade dos dispositivos. o Descrição da fonte de alimentaçãoCabeça do formulário | Apresentar um pedido de informações pormenorizadas sobre o dispositivo. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.6 | Labelling proposta | Forneça as Instructions for Use (IFU), o Manual do Utilizador e qualquer material promocional associado ao dispositivo ativo. | Apresentar um pedido de Instructions for Use (IFU), Manual do Utilizador e qualquer material promocional, se disponível. Analisar as Instruções de Utilização, o Manual do Utilizador e o material promocional fornecido pelo candidato. Documentar as Instruções de Utilização, o Manual do Utilizador e o material promocional para efeitos de apresentação de 510(k). |
| 2.7 | Embalagem e transporte | Fornecer os planos de estudo e os relatórios para a validação da embalagem e do transporte. | Apresentar um pedido de plano de estudo e relatórios relativos à validação da embalagem e do transporte. Rever os planos e relatórios de estudo para a validação da embalagem e do transporte e fornecer quaisquer correcções ou comentários necessários. |
| 2.8 | Esterilização (se a esterilidade for aplicável) | Fornecer os planos e relatórios de estudo para a validação da esterilização. | Apresentar uma requisição para o plano de estudo e relatórios da Validação da Esterilização. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.9 | Teste de desempenho _ Banco de dados | Iniciar um pedido formal para os planos e relatórios do estudo de viabilidade dos testes de desempenho, descrevendo os requisitos e objectivos específicos a abordar | Apresentar uma requisição para os planos e relatórios do estudo de bancada do dispositivo ativo para ensaio de desempenho. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
Compatibilidade electromagnética e segurança eléctrica Documentação de apoio | |||
| 2.10 | Caraterísticas dos dispositivos relacionados com a CEM e ambientes de utilização previstos | Fornecer pormenores sobre as caraterísticas dos dispositivos relacionados com a CEM e os ambientes de utilização previstos, incluindo: o Uma visão geral do dispositivo. o Funções e modos de funcionamento. o Diagramas de blocos. o Fotografias, cabos e acessórios relevantes. o Interoperabilidade dos dispositivos. o Descrição da fonte de alimentação, incluindo a viabilidade de utilizar o dispositivo médico alimentado internamente durante o carregamento. o Ambientes em que o dispositivo médico se destina a ser utilizado. o Descrição de qualquer tecnologia sem fios (se aplicável) para considerações adicionais relativamente a dispositivos médicos com fios. o Descrição de quaisquer emissores de RF internos no dispositivo médico que possam potencialmente causar perturbações electromagnéticas. o Abordar os emissores electromagnéticos (EM) comuns, bem como os emissores médicos únicos.
| Apresentar uma requisição de informações sobre as caraterísticas dos dispositivos relacionados com a CEM e os ambientes de utilização previstos. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.11 | Avaliação dos riscos | Forneça um Plano de Gestão de Risco inclua uma avaliação de risco demonstrando a mitigação eficaz do risco, juntamente com um relatório abrangente de gestão de risco que englobe todos os elementos de risco. Fornecer o documento revisto com sugestões de correcções e melhorias | Envie uma requisição para o Arquivo de Gestão de Risco e solicite a documentação do Plano de Gestão de Risco Relatório, incluindo a identificação dos perigos de risco, avaliação de risco e demonstração da mitigação de risco adequada. O Relatório de Gestão de Risco deve abranger todos os elementos de risco, de preferência com secções separadas para maior clareza. Fornecer um modelo do Plano de Gestão de Risco do Relatório de Gestão de Risco que abranja todos os riscos relacionados ao dispositivo, mediante solicitação do requerente. Rever os dados do dossier de gestão dos riscos, incluindo o plano e o relatório partilhados pelo requerente, e dar sugestões para as correcções necessárias para garantir uma documentação completa para a apresentação do 510(k). Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.12 | Norma de consenso | Fornecer confirmação das normas de consenso relevantes e uma explicação de quaisquer desvios em relação às normas FDA. | Apresentar uma requisição para as normas consensuais aplicáveis relacionadas com a CEM e a segurança eléctrica para o dispositivo ativo. Documentar as normas de consenso confirmadas para o dispositivo ativo para efeitos de apresentação de 510(k). |
| 2.13 | Desempenho essencial e critérios de aprovação/reprovação de imunidade | Apresentar o plano de estudo e os relatórios dos testes de desempenho essencial e de imunidade realizados no dispositivo ativo, em conformidade com as normas FDA. | Apresentar uma requisição para o plano de estudo e relatórios de testes de Desempenho Essencial e Imunidade realizados no dispositivo ativo, de acordo com as normas FDA. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.14 | Configuração de dispositivos médicos e funções testadas | Fornecer a Configuração do Dispositivo Médico e as Funções Testadas para o dispositivo ativo, incluindo os seguintes pormenores: o Fornecer uma descrição exaustiva do dispositivo médico em teste, incluindo informações pormenorizadas sobre a sua configuração, funções, modos e as definições específicas que foram testadas. o A descrição do dispositivo em teste deve incluir o nome do dispositivo médico, o número do modelo e indicar se o dispositivo é o dispositivo médico pronto para produção final atualmente em análise. | Apresentar um pedido para as funções de configuração e teste do dispositivo médico ativo. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
| 2.15 | Resultados dos ensaios de CEM | Fornecer o plano de estudo e o relatório do teste de EMC (Compatibilidade Electromagnética) em conformidade com a norma de consenso FDA recomendada para o dispositivo ativo. | Iniciar um pedido formal para o plano e relatório do estudo de testes EMC, em conformidade com a norma de consenso FDA recomendada para dispositivos activos. Rever minuciosamente e documentar meticulosamente todas as informações necessárias em preparação para a apresentação do 510(k). |
Fase 3 - Documentos iniciais e documentos de equivalência substancial | |||
|---|---|---|---|
Requisitos do documento | Âmbito de aplicação do requerente 510(k) | Âmbito de aplicação de Freyr | |
| 3.1 | Folha de rosto da submissão de análise prévia à comercialização CDRHFDA Formulário 3514FDA ) | - | Preencher o formulário 3514 FDA utilizando os dados fornecidos pelo requerente |
| 3.2 | Resumo e certificação da classe III | - | Esta etapa não é necessária se não forem necessários estudos clínicos |
| 3.3 | Certificação financeira ou declaração de informação | - | Esta etapa não é necessária se não forem necessários estudos clínicos |
| 3.4 | Resumo executivo | - | Desenvolva um modelo e prepare meticulosamente o documento. Apresentar justificações para quaisquer discrepâncias observadas entre o dispositivo proposto e o dispositivo de referência. Estudo comparativo entre o dispositivo proposto e o dispositivo predicado escolhido, criação de um modelo e preparação do documento correspondente. |
| 3.5 | Discussão sobre equivalência substancial | - | Desenvolva um modelo e prepare meticulosamente o documento. Estudo comparativo entre o dispositivo proposto e o dispositivo predicado escolhido, criação de um modelo e preparação do documento correspondente. |
Fase 4 - Documentos administrativos | |||
|---|---|---|---|
Requisitos do documento | Âmbito de aplicação do requerente 510(k) | Âmbito de aplicação de Freyr | |
| 4.1 | Carta de acompanhamento 510(k) | Assine o documento impresso em papel timbrado da empresa e providencie o envio de uma cópia impressa por correio para o US . Fornecer uma cópia digital da carta de acompanhamento 510(k) assinada para inclusão na documentação 510(k) | Preparar um modelo completo que inclua todos os pormenores necessários para a carta de acompanhamento e fornecê-lo ao candidato. Instruir o candidato a utilizar o seu papel timbrado oficial e a certificar-se de que a carta de acompanhamento é assinada por uma pessoa autorizada |
| 4.2 | Declaração de veracidade e exatidão | Assegurar que o documento é assinado pela pessoa de contacto designada na empresa e fornecido em conformidade. | Desenvolver um modelo abrangente que contenha todo o conteúdo necessário a incluir no documento de apresentação. |
| 4.3 | Declarações de conformidade e relatório de síntese | Assegurar que o documento é assinado pela pessoa de contacto designada na empresa e fornecido em conformidade. | Desenvolver um modelo abrangente para listar e preparar sistematicamente os documentos necessários. |
| 4.4 | MDFUSCFDA Formulário 3601FDA ) | Apresentar o pagamento exigido à FDA antes da apresentação formal do dossier 510(k) | Gerar uma folha de rosto da taxa de utilização e um número de identificação pessoal (PIN) único especificamente para a apresentação do dispositivo médico. |
Fase 5 - Lista de controlo RTA e cópia eletrónica | |||
|---|---|---|---|
Requisitos do documento | Âmbito de aplicação do requerente 510(k) | Âmbito de aplicação de Freyr | |
| 5.1 | Lista de controlo do RTA | Aprovação da lista de verificação da RTA (Ready to Accept), indicando que todos os requisitos foram cumpridos com êxito | Desenvolver um modelo de lista de verificação RTA personalizado, adaptado ao tipo específico de apresentação. Complete a lista de verificação preenchendo meticulosamente todos os campos obrigatórios e assegurando que os documentos mencionados são devidamente apresentados à FDA e partilhados com o requerente. |
| 5.2 | E-Copy | Aprovação da documentação contida no dossier de apresentação final, indicando a sua conformidade com todos os requisitos e normas necessários. | Organizar as secções da pasta de apresentação de acordo com as orientações FDA e partilhá-las prontamente com o requerente. Converter a pasta de apresentação numa cópia eletrónica para facilitar o acesso e a revisão. Envie a cópia eletrónica da submissão ao US designado US . |
Registo de Dispositivos Médicos
- EstratégiaFDA abrangenteFDA US
- Identificação do dispositivo predeterminado
- Estabelecimento de equivalência substancial com o dispositivo antecedente
- Análise de lacunas paraFDA US
- Compilação de 21 secções do processo técnico 510(k)
- Publicação e criação de eCopy
- Validação e apresentação da cópia eletrónica
- Serviços de ligação para a aprovação de dispositivos
- Abordagem das respostas e deficiências do RTA
- Serviços de consultoria para resolver deficiências
- Listagem de dispositivos e manutenção da base de dados FURLS

- Trataram de muitos registos 510(k) de categorias diversificadas de dispositivos
- Equipa especializada na compilação 510(k) de acordo com os requisitos da NotificaçãoFDA (510(k))FDA US
- Apoio adicional para tratar de consultas 510(k)
- Recomendações sobre o tipo adequado de 510(k) de acordo com os requisitos de submissãoFDA (k)FDA US para o dispositivo
- Apresentação atempada dos resultados
- Em conformidade com asFDA alteraçõesFDA US
