


Registo de Dispositivos Médicos na Indonésia – Visão Geral
A Indonésia iniciou os cuidados de saúde universais para os seus cidadãos em 2014. Isto influenciou grandemente o crescimento do mercado de Dispositivos Médicos e levou ao aumento da importação de Dispositivos Médicos. Os dispositivos na Indonésia são regulados pela Agência Nacional de Controlo de Medicamentos e Alimentos (NADFC), que funciona sob a alçada do Ministério da Saúde (MoH) indonésio. O mais recente regulamento em vigor para a importação de Dispositivos Médicos é o Decreto n.º 62, imposto no ano de 2017. As empresas estrangeiras devem nomear um representante local autorizado na Indonésia para o processo de Registo de Dispositivos Médicos na Indonésia.
![]()
Autoridade Regulamentar: Agência Nacional de Controlo de Medicamentos e Alimentos (NADFC)![]()
Regulamento: N.º 62 / 2017![]()
Representante Autorizado: Representante Local Autorizado na Indonésia![]()
Requisito QMS: ISO 13485:2016![]()
Avaliação de Dados Técnicos: NADFC![]()
Validade da Licença: 5 Anos![]()
Requisitos de Rotulagem: N.º 62 / 2017![]()
Formato de Submissão: Online/Papel![]()
Idioma: Inglês e Indonésio
Classificação de Dispositivos Médicos na Indonésia
A regulamentação atual classifica os dispositivos como A, B, C e D com base no risco.
| Critérios de Risco | Classe do Dispositivo |
|---|---|
| Baixo Risco | A |
| Risco Baixo a Moderado | B |
| Risco Moderado a Elevado | C |
| Alto Risco | D |
Representante Local Autorizado na Indonésia
Os regulamentos indonésios exigem que os fabricantes nomeiem um representante local com uma Licença de Distribuidor. Pode ser nomeado um distribuidor para representar o fabricante estrangeiro na Indonésia. No entanto, a nomeação de um terceiro independente proporcionaria flexibilidade para mudar de distribuidores ou nomear vários distribuidores para uma melhor penetração no mercado.
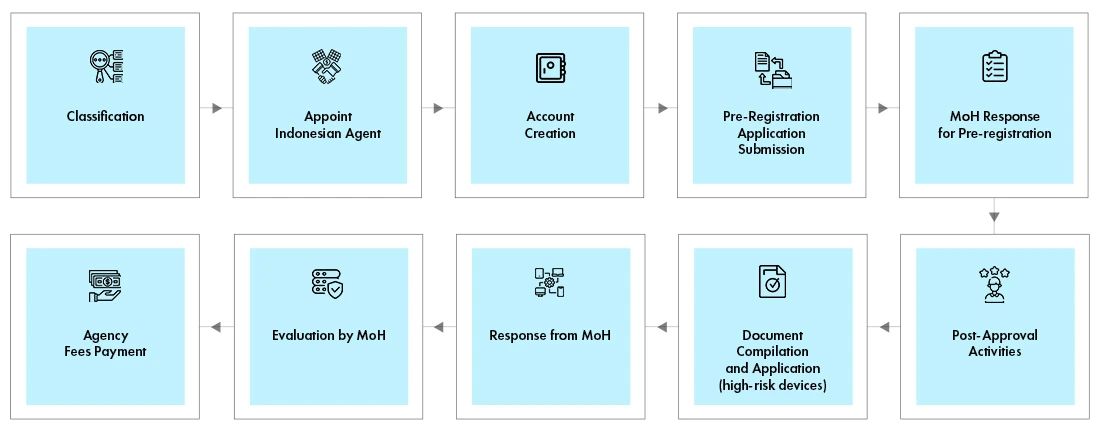
Registo de Dispositivos Médicos na Indonésia
O Representante Local deve criar uma conta no portal online. O processo de registo é o mesmo para todas as classes de dispositivos. No entanto, o requisito de documentação varia com a classe do dispositivo. O registo é um processo de duas fases –
- Processo de Pré-Registo
- Processo de Avaliação
O MoH verifica a classificação do dispositivo e determina o custo da avaliação. O resultado do pré-registo, juntamente com a fatura, é enviado por e-mail ao requerente. O representante local, em nome do fabricante, efetuará o pagamento e carregará o comprovativo de pagamento. O MoH analisará os documentos e partilhará os resultados por e-mail com o requerente. Alguns dispositivos exigem testes no país num Laboratório acreditado.
Visão Geral do Processo de Aprovação Regulamentar
A equipa de especialistas da Freyr acompanha as tendências e regulamentações em constante mudança e ajuda as partes interessadas a manter a conformidade regulamentar ao longo do ciclo de vida do produto. Oferecemos soluções de Regulamentação para manter outros aspetos de conformidade regulamentar dentro de orçamentos limitados.
Classe do Dispositivo | Classe de Risco | Prazos para o MoH Autorização de Introdução no Mercado | Prazos para o MoH Renovação / Variação | ||
|---|---|---|---|---|---|
| Processo de Classificação (Dias) | Processo de Avaliação (Dias) | Processo de Classificação (Dias) | Processo de Avaliação (Dias) | ||
| Classe A | Baixo Risco | 7 | 45 | 7 | 45 |
| Classe B | Risco Baixo a Moderado | 7 | 90 | 7 | 45 |
| Classe C | Risco Moderado a Elevado | 7 | 100 | 7 | 45 |
| Classe D | Alto Risco | 7 | 120 | 7 | 45 |
Experiência Freyr
- Diligência Devida Regulamentar
- Registo de Dispositivos
- Testes no País
- Licenciamento de Distribuidores.
- Legalização e Autenticação Notarial
- Representante Legal
- Apoio à rotulagem
- Apoio à Tradução
- Identificação e qualificação de distribuidores
- Serviços de Vigilância Pós-Comercialização
- Gestão de Alterações Pós-Aprovação
- Serviços de renovação e transferência de licença
- Serviços de submissão e ligação
