


Visão Geral do Registo de Dispositivos Médicos em Taiwan
Taiwan tem uma procura crescente por Dispositivos Médicos. A Taiwan Food & Drug Administration (TFDA), sob a alçada do Ministério da Saúde e Bem-Estar (MOHW), regulamenta os Dispositivos Médicos através da Lei dos Assuntos Farmacêuticos (PAA). Fabricantes estrangeiros sem escritório físico em Taiwan necessitam de representação de um agente em Taiwan como pré-requisito para o processo de Registo de Dispositivos Médicos em Taiwan.
![]()
Autoridade Regulamentar: Taiwan Food and Drug Administration![]()
Regulamento: Lei de Assuntos Farmacêuticos (PAA) e Regulamento para o Registo de Dispositivos Médicos![]()
Representante Autorizado: Representação de Agente em Taiwan necessária![]()
Requisito QMS: Documentação do Sistema de Qualidade (QSD) ISO 13485![]()
Avaliação de Dados Técnicosa: Divisão de Dispositivos Médicos e Cosméticos![]()
Validade da Licença: QSD - 3 Anos; Registo de Produto - 5 Anos![]()
Requisitos de Rotulagem: Artigo 75, Lei dos Assuntos Farmacêuticos![]()
Formato de Submissão: Documento![]()
Idioma: Inglês e Chinês
Classificação de Dispositivos Médicos em Taiwan
A TFDA classifica os Dispositivos Médicos em 3 classes com base no risco: Classe I para baixo risco, Classe II para risco moderado e Classe III para dispositivos de alto risco. A necessidade de um dispositivo precedente representa um desafio para a entrada de dispositivos inovadores no mercado. O aumento do tempo de procedimento para dispositivos das Classes II e III que necessitam de Documentação do Sistema de Qualidade é outra complexidade envolvida. Todos os Dispositivos Médicos importados devem obter um certificado de registo da TFDA.
| Classe do Dispositivo | Risco |
|---|---|
| Classe I | Baixo Risco |
| Classe II | Risco Moderado |
| Classe III | Alto Risco |
Representação de Agente em Taiwan
Fabricantes estrangeiros sem escritório físico em Taiwan devem nomear um Agente em Taiwan como pré-requisito para comercializar dispositivos em Taiwan. Nomear uma organização terceira como Agente em Taiwan, em vez de um distribuidor, oferece flexibilidade para explorar múltiplos distribuidores para uma melhor penetração no mercado. O Agente em Taiwan deve ter uma entidade legal estabelecida em Taiwan, certificada com uma Licença de Vendas Farmacêuticas.
Registo de Dispositivos Médicos em Taiwan
Antes que um Dispositivo Médico possa ser vendido em Taiwan, o registo da Documentação do Sistema de Qualidade (QSD) para a instalação de fabrico é exigido, além do registo do Dispositivo Médico. O registo QSD é dispensado apenas para Dispositivos Médicos de Classe I (não estéreis). Uma licença QSD (recebida após a aprovação do registo QSD) em Taiwan é semelhante às Boas Práticas de Fabrico (BPF) para Dispositivos Médicos.
A TFDA anunciou que, a partir de 1 de junho de 2022, os titulares de licenças de dispositivos médicos de Classe III são obrigados a carregar o UDI e as informações correspondentes do produto para a Base de Dados UDI (UDID). Os fabricantes de dispositivos médicos também são obrigados a colocar o UDI na etiqueta do produto. Além disso, a partir de 1 de junho de 2023, os dispositivos médicos de Classe II são obrigados a cumprir os regulamentos relevantes do UDI.
Fluxo do processo
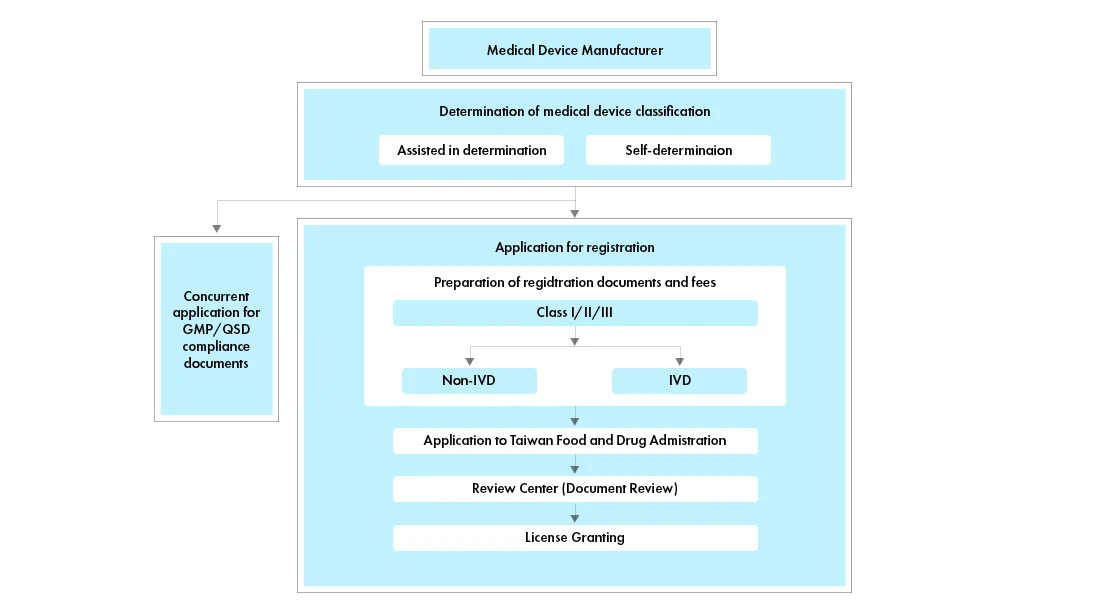
Gestão do Ciclo de Vida de Dispositivos Médicos Pós-Aprovação
A Freyr apoia os fabricantes estrangeiros na gestão End-to-End do ciclo de vida de dispositivos médicos, incluindo atividades pós-aprovação, tais como:
- Gestão de alterações pós-aprovação - modificações a aprovações de Dispositivos Médicos existentes, como a adição de novas variantes, acessórios; adição de novas indicações de utilização, entre outros
- Manutenção de aprovações e registo através do pagamento atempado de taxas administrativas e de registo
- Renovação de licenças
- Intermediação entre a TFDA e o fabricante
- Gestão de Importações
Freyr especializa-se em atender às necessidades Regulamentares de Dispositivos Médicos em Taiwan. Com uma vasta rede, Freyr ajuda na nomeação de um agente local fiável cuja presença é de extrema importância durante toda a vigilância pós-comercialização. Os nossos especialistas também auxiliam na seleção de um dispositivo de referência adequado e aprovações existentes de outros mercados para apoiar a entrada de novos dispositivos no mercado.
Resumo
| Classe do Dispositivo | Critérios de Risco / Classificação | QMS | Registo de produtos |
|---|---|---|---|
| Classe I | Baixo Risco | Isentos (dispositivos não estéreis de Classe I) | Sim |
| Classe II | Risco Moderado | QSD | Sim |
| Classe III | Alto Risco | QSD | Sim |
Experiência Freyr
- Diligência Devida Regulamentar
- Classificação Oficial
- Aprovações QSD
- Registo de Dispositivos
- Representante Legal
- Apoio à rotulagem
- Apoio à Tradução
- Identificação e qualificação de distribuidores
- Vigilância Pós-Comercialização
- Gestão de Alterações Pós-Aprovação
- Renovação e transferência de licenças
- Submissão e coordenação
