


Visão Geral do Registo de Dispositivos Médicos na Turquia
O mercado turco de dispositivos médicos tem registado um crescimento significativo e consistente na última década. A partir de 2021, o Registo de Dispositivos Médicos na Turquia exige o cumprimento do Regulamento (UE) 2017/745 relativo aos Dispositivos Médicos (MDR) e do Regulamento (UE) 2017/746 relativo aos Dispositivos Médicos para Diagnóstico In Vitro (IVDR). Isto impulsionou o comércio internacional, levando várias empresas globais a lançar os seus dispositivos médicos no país.
![]()
Autoridade Regulamentar: Agência Turca de Medicamentos e Dispositivos Médicos (TITCK)![]()
Regulamento: Regulamento dos Dispositivos Médicos (MDR) 2017/745, Regulamento dos Dispositivos Médicos para Diagnóstico In Vitro 2017/746![]()
Via Regulamentar: A marcação CE é obrigatória, seguida de Registo/Notificação no Sistema de Rastreio de Produtos (UTS)![]()
Representante local na Turquia![]()
Requisito QMS: ISO 13485:2016![]()
Avaliação de Dados Técnicos: Organismo Notificado para a marcação CE![]()
Validade da Licença: Ilimitada![]()
Formato de Submissão: Documento![]()
Tradução: Documentos Traduzidos para Turco
Classificação de Dispositivos Médicos
A Turquia segue a mesma classificação de dispositivos médicos que a estabelecida no EU MDR e IVDR. Determinar a classificação do dispositivo pode ser um desafio e, por isso, ter o apoio de um consultor regulamentar experiente é bastante crucial aqui.
Classes de Dispositivos Médicos
| Classe | Risco |
|---|---|
| Classe I | Baixa |
| Classe IIa | Moderado |
| Classe IIb | Moderado a Alto |
| Classe III | Elevada |
Classes de dispositivos de diagnóstico in vitro
| Classe | Risco |
|---|---|
| Classe A | Baixa |
| Classe B | Moderado |
| Classe C | Moderado a Alto |
| Classe D | Elevada |
Registo de Dispositivos Médicos
A marcação CE é uma conformidade exigida aos fabricantes para colocar o seu dispositivo no mercado da Turquia. A marcação CE é emitida através de uma avaliação de conformidade realizada pelo organismo notificado. Atualmente, a Turquia está autorizada a nomear organismos notificados de acordo com o EU MDR e IVDR.
As empresas são obrigadas a registar-se no Sistema de Registo Central (MERSIS) e a registar o dispositivo no Sistema de Rastreamento de Produtos (UTS).
Fluxo do processo
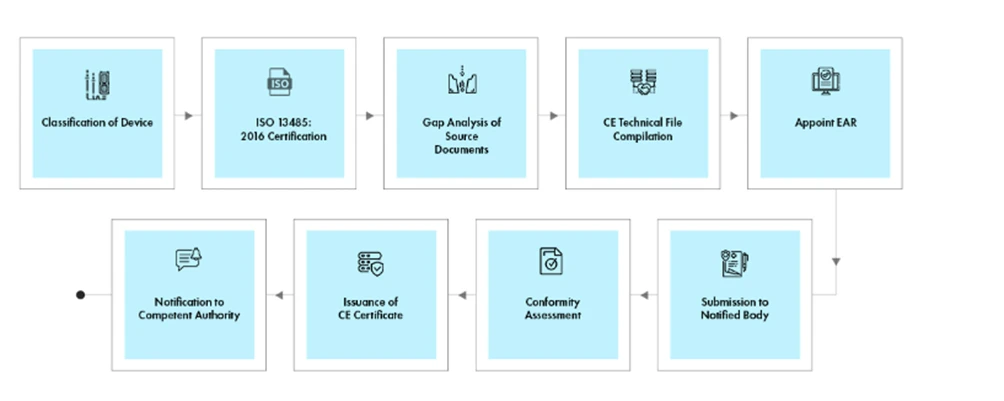
Gestão do Ciclo de Vida do Dispositivo Pós-aprovação
A Freyr apoia fabricantes estrangeiros na gestão End-to-End do ciclo de vida de dispositivos médicos, incluindo atividades pós-aprovação, tais como:
- Gestão de alterações pós-aprovação - modificações a aprovações existentes de dispositivos médicos, tais como a adição de novas variantes, acessórios; adição de novas indicações de uso, entre outros
- Manutenção da ISO 13485:2016 e da certificação CE
- Renovação de licenças
- Ligação entre o Organismo Notificado e o fabricante
Com várias entidades de autorização envolvidas, os fabricantes estrangeiros precisam de cumprir múltiplos conjuntos de regulamentos em cada processo individual para aprovações de dispositivos. A obtenção de uma marcação CE e a posterior adesão a regulamentos específicos de cada estado exige um conhecimento regulamentar extenso. Por vezes, sem um parceiro regulamentar comprovado, navegar por todos os requisitos dos dispositivos pode ser um desafio para os novos participantes no mercado. Para ajudar os fabricantes, a Freyr fornece serviços regulamentares End-to-End para acelerar as aprovações de dispositivos médicos.
Experiência Freyr
- Classificação Europeia de Dispositivos Médicos
- Apoio ao Representante Autorizado Europeu (EAR)
- Registo de Dispositivos e Notificação de Produtos na Turquia
- Consulta sobre Gestão de Risco ISO 14971:2019
- Conformidade com a ISO 13485:2016
- Revisão, compilação e submissão do ficheiro técnico CE/dossiê de conceção.
- Apoio à Transição para o EU MDR
- Apoio à Transição para o Regulamento IVDR da UE
- Relatórios de Avaliação Clínica (CER) para Dispositivos Médicos
- Relatórios de Avaliação de Desempenho (PER) para Dispositivos Médicos para Diagnóstico In Vitro
- Notificação/Registo de Dispositivos Médicos através do Sistema de Registo online
- Relatório de estratégia regulamentar de Dispositivos Médicos
- Apoio a testes – biocompatibilidade, segurança elétrica, mecânicos e de desempenho
- Apoio à Conformidade da Rotulagem
- Apoio GMP
- Apoio à Vigilância Pós-Comercialização
